Article Text

Abstract
Background There are few therapeutic options available for patients with B-cell acute lymphoblastic leukemia (B-ALL) relapsing as CD19– either after chemotherapy or CD19-targeted immunotherapies. CD22-chimeric antigen receptor (CAR) T cells represent an attractive addition to CD19-CAR T cell therapy because they will target both CD22+CD19– B-ALL relapses and CD19– preleukemic cells. However, the immune escape mechanisms from CD22-CAR T cells, and the potential contribution of the epitope binding of the anti-CD22 single-chain variable fragment (scFv) remain understudied.
Methods Here, we have developed and comprehensively characterized a novel CD22-CAR (clone hCD22.7) targeting a membrane-distal CD22 epitope and tested its cytotoxic effects against B-ALL cells both in in vitro and in vivo assays.
Results Conformational epitope mapping, cross-blocking, and molecular docking assays revealed that the hCD22.7 scFv is a high-affinity binding antibody which specifically binds to the ESTKDGKVP sequence, located in the Ig-like V-type domain, the most distal domain of CD22. We observed efficient killing of B-ALL cells in vitro, although the kinetics were dependent on the level of CD22 expression. Importantly, we show an efficient in vivo control of patients with B-ALL derived xenografts with diverse aggressiveness, coupled to long-term hCD22.7-CAR T cell persistence. Remaining leukemic cells at sacrifice maintained full expression of CD22, ruling out CAR pressure-mediated antigen loss. Finally, the immunogenicity capacity of this hCD22.7-scFv was very similar to that of other CD22 scFv previously used in adoptive T cell therapy.
Conclusions We report a novel, high-affinity hCD22.7 scFv which targets a membrane-distal epitope of CD22. 4-1BB-based hCD22.7-CAR T cells efficiently eliminate clinically relevant B- CD22high and CD22low ALL primary samples in vitro and in vivo. Our study supports the clinical translation of this hCD22.7-CAR as either single or tandem CD22–CD19-CAR for both naive and anti-CD19-resistant patients with B-ALL.
- cell engineering
- hematologic neoplasms
- immunotherapy
- receptors, chimeric antigen
- T-lymphocytes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
B-cell acute lymphoblastic leukemia (B-ALL) is an aggressive cancer, diagnosed at any age throughout the lifespan of an individual, being the most common malignancy in children.1 Despite current 5-year disease-free survival rates of ~80%, refractory and relapsed (R/R) patients have a dismal prognosis.2–7 Adult B-ALL is less frequent, but is commonly associated with an unfavorable clinical outcome even after the use of high-dose chemotherapy regimens and allogenic stem cell transplantation.8
Immunotherapy has generated unprecedented expectations in cancer treatment. Adoptive cellular immunotherapy, based on the genetic engineering of human T cells with chimeric antigen receptors (CARs) redirected against cell surface tumor antigens, has shown great potential in R/R B-ALL due to the high efficacy and specificity of CD19-CAR T cells, with ~90% response rates and ~50% complete remission rates after 12 months.9 10 However, CD19-CAR T cells are not always curative and ~60% of patients inevitably relapse, mainly because of poor CAR T cell persistence or the emergence of CD19– B-ALL clones.11 12 Of note, a very recent and elegant clinical study has shown that a low-affinity CD19-CAR enhances CAR T cell expansion and persistence in patients with B-ALL, but it is associated with immune escape and an increased number of CD19– relapses.12 Unfortunately, there are very few therapeutic options for patients with B-ALL relapsing as CD19– after either chemotherapy or CD19-directed immunotherapy and, therefore, novel strategies are being explored to enhance CAR T cell function and persistence, including the development of novel target antigens, costimulatory domains or effector cells.13
Like CD19, CD22 is expressed in a B-cell lineage-restricted fashion.14 CD22-directed T cells are an attractive addition to the current CD19-directed T cell strategies because (1) they constitute an alternative for CD19–/dim B-ALL relapses as CD22 is retained in such relapses,15 and (2) they would target the earliest pre-VDJ CD19–B-lineage progenitors, which potentially can escape in CD19-CAR T cell therapy.16 Indeed, CD22 has been employed as a target for B-ALL in clinical trials using anti-CD22 monoclonal antibodies (moAb) linked to immunotoxins,17–19 and several CD22-CARs have also been reported.15 20–22 CD22-CAR T cells induce clinical remission in ~70% of patients with R/R B-ALL either naive or resistant to CD19-CAR T cells.15 21 23 Moreover, these studies reported somewhat different clinical results regarding the potential mechanism of immune escape of B-ALL blasts from CD22-CAR T cells-mediated lysis with subsequent CD22–/dim relapses.15 21 Importantly, comparative studies from the Orentas laboratory suggested that maximal efficacy of CD22-CAR T cells is obtained when using anti-CD22 antibodies that target cell surface proximal epitopes or that display a high-binding affinity for CD22.20 Here, we have developed and characterized a novel high-affinity CD22-CAR targeting a membrane-distal CD22 epitope, and report an efficient elimination in vitro and in vivo of clinically relevant primary B-ALL cells.
Materials and methods
Generation of a CD22 single-chain variable fragment (scFv)
MoAbs reactive with human CD22 were generated by fusion of NS-1 myeloma cells with splenocytes from a Balb/c mouse previously immunized three times with 30×106 CD22-transfected 300.19 cells. Supernatants from hybridoma-containing wells were screened by flow cytometry for moAbs reactive with CD22-transfected 300.19 cells. Three reactive hybridomas were chosen for further characterization and were subcloned by limiting dilution (clones hCD22.7, hCD22.316 and hCD22.401). The three antibodies were IgG1κ isotype, as determined using a mouse moAb isotyping kit (Boehringer Mannheim, Mannheim, Germany). Antibody specificity was further validated using a CD22-transfected COS cell line (pUNO1-hCD22; InvivoGen, Toulouse, France), the cell lines Raji, Daudi, PRMI 82229, U266 and Jurkat, and peripheral blood (PB) mononuclear cells (PBMCs). CD22 epitope binding to the hCD22.7 clone was determined by cross-blocking24 with an antibody against the N-terminal domain of CD22 (S-HCL-1).25 The CD22-specific scFv derived from the hCD22.7 clone was obtained using the mouse IgG Library Primer Set (Progen, Heidelberg, Germany).
Immunogenicity prediction
Immunogenicity potential was predicted using the in silico tool NetMHC V.4.0.26 27 Analyzed scFvs were broken in 9-mer peptides, length for which human leukocyte antigen (HLA) molecules have a strong preference. Peptides were considered to bind major histocompatibility complex class I (MHC-I) when a half-maximum inhibitory concentration (IC50) is <500 nM, being considered strong binders when IC50 <50 nM. Only HLA supertype representatives were analyzed. The predicted epitope-binding affinity was calculated as 1/IC50 100.28
Epitope mapping
Reactivity of the hCD22.7 moAb was subjected to PEPperMAP Conformational Epitope Mapping against CD22 translated into cyclic constrained peptides (PEPperPRINT GmbH, Heidelberg, Germany). The sequence of CD22 (UniProtKB P20273) was elongated with neutral GSGSGSG linkers at the C-terminus and N-terminus to avoid truncated peptides. The elongated antigen sequence was translated into 7, 10 and 13 amino acid peptides with a peptide–peptide overlap of 6, 9 and 12 amino acids. After peptide synthesis, all peptides were cyclized via a thioether linkage between a C-terminal cysteine and an appropriately modified N-terminus. The resulting conformational CD22 peptide microarrays contained 2556 different peptides printed in duplicate (5112 peptide spots), and were framed by additional human influenza hemagglutinin (HA, YPYDVPDYAG, 146 spots) control peptides. Hibridoma supernatant containing hCD22.7 moAb was diluted in incubation buffer (phosphate-buffered saline (PBS), pH 7.4, with 0.005% Tween 20) and 10% MB-070 blocking buffer (Rockland Immunochemicals, Limerick, Pennsylvania, USA) and incubated with peptide microarrays for 16 hours at 4°C and swirling at 140 rpm. Unbound antibodies were removed by washing the arrays with washing buffer (PBS, pH 7.4, with 0.005% Tween 20). Prestaining of the peptide arrays was done with 0.2 µg/mL goat anti-mouse IgG (H+L) antibody conjugated to DyLight680 (Thermo Fisher Scientific). HA control peptides framing the peptide arrays were finally stained with 0.5 µg/mL monoclonal anti-HA (12CA5)-DyLight800 (Thermo Fisher Scientific) for 45 min at room temperature (RT). Quantification of spot intensities and peptide annotation was performed with the LI-COR Odyssey Imaging System (scanning offset 0.65 mm, resolution 21 µm, and scanning intensities of 7/7 (red=700 nm/green=800 nm) and PepSlide Analyzer.
Structural model
The structural model of the CD22-hCD22.7-scFv complex was obtained using directed docking as follows. The structure for CD22 was taken for the PDB databank29 (identification code 5vkj30) and the structure of the scFv anti-CD22 was derived by homology modeling using M4T31 and templates 4h0h,32 1dqd33 and 4okv.34 The initial docking conformations were inferred using PatchDock35 by selecting the complementarity-determining regions of anti-CD22-scFv and mapped epitope of CD22 as docking interfaces. Resulting docking conformations were further refined and minimized using Rosetta36 and the top 200 poses were visually inspected. A more detailed description of the structural analysis is shown in online supplementary information.
Supplemental material
CAR design and vectors, lentiviral production and T cells transduction
The CD22-specific scFv was cloned into a pCCL lentiviral-based backbone containing a human CD8 hinge and transmembrane (TM) domain, human 4-1BB and CD3ζ endodomains (second-generation CAR), and a T2A-GFP cassette.37 An identical lentiviral vector with the CD8 TM-4-1BB-CD3ζ domains linked to a His-Tag was used as a mock intracellular (mock-IC) control. CAR-expressing viral particles pseudotyped with VSV-G were generated by cotransfection of HEK 293 T cells with the pCCL vector and the packaging plasmids VSV-G and psPAX2 using polyethylenimine (PEI, Polysciences, Warrington, Pennsylvania, USA). Supernatants were collected at 48 and 72 hours after transfection and concentrated by ultracentrifugation.
PBMCs were isolated from buffy coats from healthy volunteers by Ficoll-Hypaque gradient centrifugation (GE Healthcare, Chicago, Illinois, USA). Buffy coats were obtained from the Barcelona Blood and Tissue Bank on Institutional Review Board approval (HCB/2018/0030). T cells were activated by plate coating with anti-CD3 (OKT3) and anti-CD28 (CD28.2) antibodies (BD Biosciences, Franklin Lakes, New Jersey, USA) for 2 days and were transduced with the CAR-expressing lentivirus at a multiplicity of infection of 10 in the presence of interleukin 7 (IL-7) and IL-15 (10 ng/mL; Miltenyi Biotec, Bergisch Gladbach, Germany).37 T cells were expanded in RPMI-1640 medium (Gibco/Invitrogen, Waltham, MA) containing 10% heat-inactivated fetal bovine serum (FBS, Sigma, St. Louis, Missouri, USA), penicillin-streptomycin (Gibco/Invitrogen), and IL-7 and IL-15 (10 ng/mL), for up to 10 days. Surface expression of CD22-CAR was traced by fluorescence-activated cell sorting (FACS).
Cell lines
SEM, NALM6, MV4-11 and REH cells lines were purchased from the DSMZ cell line bank (Braunschweig, Germany). CD19-knock out (KO) and CD22-KO SEM cells were generated by CRISPR-mediated genome editing. Briefly, 200,000 cells were electroporated (Neon transfector, ThermoFisher Scientific, Waltham, Massachusetts, USA) with a Cas9 protein/tracrRNA/crRNA complex (IDT, Coralville, Iowa, USA). Two guides were designed for each gene: CD19-exon 2.1, CAGGCCTGGGAATCCACATG and CD19-exon 14.1, AGAACATGGATAATCCCGAT; and CD22-exon 3.2, TCAATGACAGTGGTCAGCTG and CD22-exon 9, CAGGTGTAGTGGGAGACGGG. After electroporation, cells were allowed to recover and then CD19– or CD22– cells were FACS sorted (>99% purity).
In vitro cytotoxicity assays and cytokine release determination
Target cells (cell lines or primary B-ALL cells; 100,000 target cells/well of a 96-well plate) were incubated with CD22-CAR or mock-IC T cells at different effector:target (E:T) ratios for the indicated time periods. Cell lines were cultured in RPMI-1640, 10% FBS and penicillin–streptomycin. Primary cells were cultured in StemSpanTM SFEM media (StemCell Technologies, Vancouver, Canada), 20% FBS, penicillin–streptomycin, insulin–transferrin–selenium (Gibco/Invitrogen), hSCF (100 ng/mL), hFLT3L (100 ng/mL), hIL3 (10 ng/mL) and hIL7 (10 ng/mL, all from Miltenyi Biotec, Bergisch Gladbach, Germany). CAR T cell-mediated cytotoxicity was determined by analyzing the residual live (7-aminoactinomycin D–; 7-AAD–) target cells at each time point and E:T ratio. For absolute cell counting, BD TruCountTM absolute count tubes (BD Biosciences) were used. Quantification of the proinflammatory cytokines IL2, TNFα and IFNγ was measured by ELISA using BD OptEIATM Human ELISA kits (BD Biosciences), in supernatants harvested at 24 hours (cell lines) and 48 hours (primary cells) post T cell-exposure, using an 1:1 E:T ratio. Online supplementary table 1 shows clinic-biological features of the B-ALL samples used.
Flow cytometry
Cell surface expression of the CD22-CAR was confirmed by green fluorescence protein (GFP) expression, binding to an AffiniPure F(ab')₂ fragment goat anti-mouse IgG (H+L)-APC (Jackson ImmunoResearch, Westgrove, Pennsylvania, USA), and an anti-His-APC (J095G46; BioLegend, San Diego, California, USA), after prior incubation with human recombinant CD22-His (rCD22-His, ThermoFisher Scientific). Activation of transduced T cells was confirmed 48 hours after plating of PBMCs by surface staining with CD3-PE (UCHT1; BD Biosciences), CD25-APC (M-A251; BD Biosciences) and CD69-VioBlue (REA824; Miltenyi Biotec). T cells and target cells were identified in in vitro assays using the following human antibodies: CD3-PE (UCHT1), CD22-APC (HIB22), CD19-BV421 (HIB19) and CD10-PECy7 (HI10a) or CD13-PECy7 (WM15, for SEM cells) or CD33-APC (WM53, for MV4-11 cells, BD Biosciences). Dead cells were discarded by 7AAD staining. Cells collected from mouse PB, bone marrow (BM) and spleen were stained with HLA-ABC-PE (G46-2.6), CD45-BV510 (HI30), CD3-PerCP (SK7), CD22-APC, CD19-BV421 and CD10-PECy7 (BD Biosciences). Cells were incubated for 30 min and then lysed and fixed with the BD FACSTM Lysing solution (BD Biosciences). Fluorescence Minus One controls were used to set the gates (online supplementary figure 1). A FACSCantoTM-II flow cytometer and a FACSDivaTM software was used for the analysis (BD Biosciences).
In vivo xenograft models for B-ALL and CAR T cells
Twelve-week-old non-obese diabetic (NOD).Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory, Bar Harbor, Maine USA) were bred and housed under pathogen-free conditions. All in vivo procedures were approved by a local ethics committee (HRH-17–0029 P1). In total, 0.5–1.5×106 patient derived xenograft (PDX) B-ALL cells were intra-BM transplanted in sublethally irradiated (2 Gy) NSG mice, followed by intravenous infusion of 4×106 CD22-CAR T cells (average transduction ~51.3%) or mock-IC T cells (average transduction ~54.2%) 2 weeks later. B-ALL engraftment was monitored in PB every other week. BM aspirates were analyzed 6 weeks after transplantation and at sacrifice, along with spleens. Red blood cell counts were determined with a hemocytometer 2800VET V-Sight (Menarini Diagnostics, Badalona, Spain).
Statistical analysis
All data are expressed as the mean±SEM. Differences between groups were assessed using two-tailed unpaired Student’s t-tests, or paired Student’s t-tests when analyzing data from different donors, unless otherway stated. All analyses were performed with Prism software, V.8.0 (GraphPad software, San Diego, California, USA).
Results
hCD22.7-CAR T cells efficiently eliminate B-ALL cells in vitro
Our novel anti-CD22 scFv (clone hCD22.7) was cloned into a second-generation CAR construct consisting of an anti-CD22 scFv, a CD8 hinge/TM spacer, and intracellular signaling domains from 4-1BB and CD3ζ, coupled in-frame with GFP through a T2A sequence (figure 1A). To identify the domain of CD22 recognized by the hCD22.7 moAb, we performed a cross-blocking assay with the anti-CD22 clone H-SCL-1, known to bind to the most distal Ig extracellular domain 1 of CD22.25 Binding of the labeled H-SCL-1 clone was hindered in cells preincubated with the antibody hCD22.7, indicating an overlapping binding of both moAbs to the most distal Ig domain of CD22 (figure 1B). Additionally, conformational epitope mapping assay, performed to exactly identify the sequence recognized by the hCD22.7 scFv, revealed that hCD22.7 scFv specifically binds to the ESTKDGKVP sequence, located in the Ig-like V-type domain, the most distal domain of CD22, thus confirming results from the cross-blocking assay (online supplementary figure 2). The molecular docking of this binding was computationally modeled (figure 1C and online supplementary file 1). The structural model inferred a CD22-hCD22.7-scFv complex energy of −1407.691 Rosetta score,36 and a binding free energy of −16.5615 kcal/mol, using the InterfaceAnalyzer.38 These binding affinity parameters were comparable, or even slightly higher than those of the clinically approved high-affinity anti-CD22 moAb Epratuzumab (−1005.624 Rosetta score and −11.345 kcal/mol, respectively).30

Design, detection and expansion of hCD22.7-CAR T cells. (A) Scheme of the hCD22.7-CAR structure and the mock-IC. (B) Histogram showing the blocking of the binding of the anti-CD22 S-HCL-1 clone (PE signal) when CD22+ B cell line Raji cells are preincubated with the hCD22.7 clone (left panel). Right panel depicts the CD22 structure, indicating the binding site of hCD22.7 scFv (as well as m971, HA22 and BL22 scFvs). (C) Structural model of the CD22-hCD22.7–scFv complex. The structure is predicted based on the identified binding epitope of CD22 (ESTKDGKVP, in red) and the sequence of the heavy and light chains of the hCD22.7 scFv (blue and pink, respectively). (D) Representative CAR detection in primary T cells. CD22-CAR-transduced T cells are detected as GFP+ and are corecognized by an anti-human IgG F(ab')2 and an anti-His moAb (on incubation with rCD22-His). (E) Proper T cell activation determined by CD25 and CD69 staining after 48 hours exposure to anti-CD3/CD28 (PBMCs from n=3 healthy donors). (F) Robust expansion of activated T cells untransduced or transduced with either mock-IC or CD22-CAR (PBMCs from n=3 healthy donors). Plot shows mean±SEM (n=3 healthy donors). CAR, CD22-chimeric antigen receptor; FSC, forward scatter; His, histidine; mock-IC, mock-intracellular; PBMCs, peripheral blood mononuclear cells; rCD22-His, recombinant CD22-His; scFv, single-chain variable fragment; TM, transmembrane.
Healthy human T cells were successfully transduced with both CD22-CAR and mock-IC lentivectors with a transduction efficiency of 20%–50%. The expression of the CD22-CAR in T cells was confirmed by codetection of scFv and GFP, and by using an anti-His moAb on incubation with human rCD22-His (figure 1D). Importantly, activated (CD69+CD25+) CD22-CAR T cells (figure 1E) continuously expanded >100-fold over a 15-day period, similar to mock-IC T cells (figure 1F), demonstrating that hCD22.7-CAR expression does not hamper T cell expansion.
CD22-CAR T cells from three different healthy donors were then functionally tested in vitro in cytotoxicity assays against the B-ALL cell lines NALM6, SEM and REH, and the AML cell line MV4-11 as a negative control, at increasing E:T ratios (figure 2A). Alive target cells after hCD22.7-CAR T cell exposure were identified as 7AAD–CD3–CD19+CD10+ (or CD13+ in the case of the SEM cells and 7AAD–CD3–CD33+ in the case of MV4-11 cells, figure 2B). Compared with mock-IC T cells, hCD22.7-CAR T cells specifically eliminated CD22+ B-ALL cells, even at relatively low E:T ratios (figure 2C). Specificity of the hCD22.7-CAR T cells was further demonstrated using CRISPR/Cas9-mediated CD19-KO and CD22-KO SEM cells (figure 2D). Furthermore, B-ALL cells barely survived exposure to hCD22.7-CAR T cells in a 48 hours absolute number assay at a 1:1 E:T ratio (figure 2E). hCD22.7-CAR T cells produced high levels of the proinflammatory cytokines IL2, TNFα, and IFNγ after coculture with target cells, confirming their cytotoxicity (figure 2F).

CD22-CAR T cells specifically target and eliminate B-ALL cell lines in vitro. (A) Experimental design of the in vitro cytotoxicity assays. (B) Flow cytometry gating strategy was used to analyze cytotoxicity. Alive target cells were identified as 7AAD–CD3–GFP–CD19+CD10+/CD13+ (CD33+ for MV4-11 cell line). CD22 was not used to avoid confounding detection of target cells due to potential antigen loss. Doublets were removed from the analysis. (C) Percentage of alive target cells after 48 hours incubation with CD22-CAR T cells at the indicated E:T ratios. Results are normalized respect to mock-IC data (PBMCs from n=3 healthy donors). CD22 expression in the different cell lines is shown previously in a histogram. (D) Specificity of CD22-CAR using CRISPR-Cas9 generated CD19-KO and CD22-KO SEM cells (PBMCs from n=3 healthy donors). CD22 expression in the different cell lines is shown previously in a histogram. (E) Absolute counts of alive target cells measured by FACS in 48 hours cytotoxicity assays at 1:1 E:T ratio (PBMCs from n=3 healthy donors). (F) ELISA showing robust secretion of proinflammatory cytokines by CD22-CAR T cells after 48 hours exposure to B-ALL and acute myeloid leukemia (AML) cell lines (upper panels) and wt, CD19-KO or CD22-KO SEM cell lines (bottom panels, PBMCs from n=3 healthy donors). Data are presented as mean±SEM (n=3 healthy donors). *p<0.05, **p<0.01, ***p<0.001. B-ALL, B-cell acute lymphoblastic leukemia; CAR, CD22-chimeric antigen receptor; E:T, effector:target ratio; FACS, fluorescence-activated cell sorting; FSC, forward scatter; FSC-A, forward scatter-area; FSC-H, forward scatter-high; mock-IC, mock intracellular; PBMCs, peripheral blood mononuclear cells; SSC, side scatter; KO, knock-out; MFI, median fluorescence intensity; wt, wild-type.
The expression level of CD22 impacts the in vitro efficiency of hCD22.7-CAR T cells in killing B-ALL primary cells
It has been reported that remissions post-CD22-CAR T cell therapy are short and are associated with reduced expression of CD22, whereas high expression of CD22 favors the activity of CD22-CAR T cells.22 Moreover, whether CD22–/dim B-ALL relapses are common after CD22-CAR T cell treatment remains controversial.15 21 Given this information, we next tested the efficacy of our CD22-CAR T cells in vitro, in nine primary B-ALL samples with variable expression levels of CD22. CD22 median fluorescence intensity (MFI) levels above or below the mean value (MFI=6752) were used to categorize primary B-ALLs as CD22high or CD22low (figure 3A and online supplementary figure 3a). Cytotoxicity was measured after 24 hours and 48 hours exposure to CD22-CAR/mock-IC T cells (figure 3B and online supplementary figure 3b). Overall, hCD22.7-CAR T cells significantly eliminated primary B-ALL blasts (figure 3C). However, while a sustained cell elimination of CD22high B-ALL blasts (n=3) by hCD22.7-CAR T cells was consistently observed over a 48 hours period (figure 3C, left panel), hCD22.7-CAR T cell-mediated killing of CD22low B-ALL blasts (n=6), although significant, was less robust and occurred in a shorter or delayed cytotoxic window (figure 3C, right panel). Nevertheless, a similar trend of robust production of the proinflammatory cytokines IL2, TNFα, and IFNγ was observed for all B-ALL primary samples, regardless the expression levels of CD22 (figure 3D), confirming an efficient CD22 recognition and killing of B-ALL primary cells by our hCD22.7-CAR T cells.
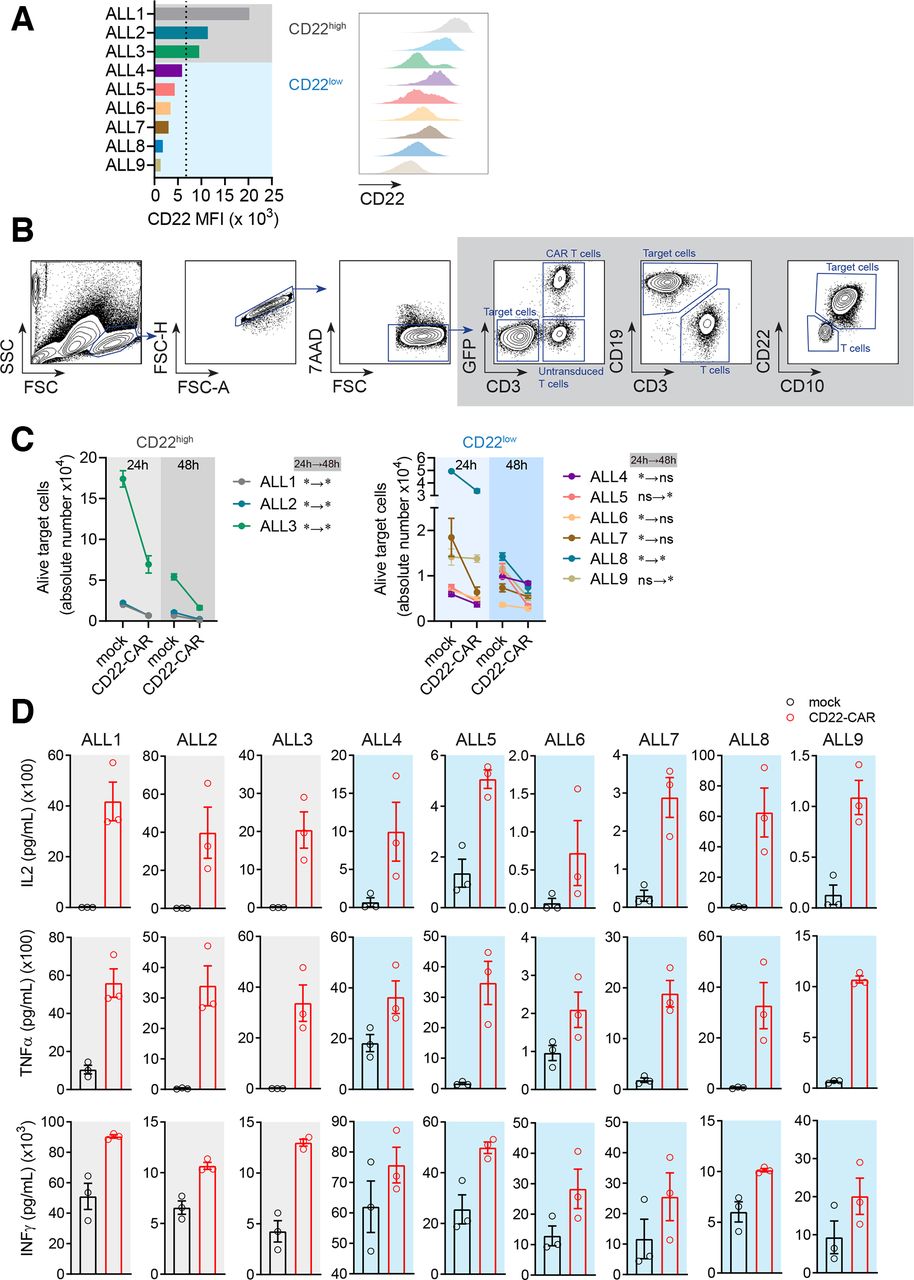
CD22-CAR T cells efficiently target and eliminate primary B-ALL cells in vitro although the expression level of CD22 impacts their efficacy. (A) MFI of CD22 in the different primary B-ALL samples used (n=9). The mean value for CD22 MFI was used to classify primary B-ALL samples as CD22high (ALL #1 to ALL #3) or CD22low (ALL #4 to ALL #9). (B) Flow cytometry gating strategy was used to analyze cytotoxicity. Alive target cells were identified as 7AAD–CD3–GFP–CD19+CD10+. CD22 was not used to avoid confounding detection of target cells due to potential antigen loss. Doublets were removed from the analysis. (C) Absolute counts of alive target CD22high and CD22low cells measured by FACS in 24 hours and 48 hours cytotoxicity assays at 1:1 E:T ratio. The statistical significance is indicated for each sample. A two-way ANOVA test (Sidak’s multiple comparison test) was used. (D) ELISA showing a similar robust production of IL2, TNFα, and IFNγ by CD22-CAR T cells after 48 hours exposure, among all B-ALL primary samples regardless the expression levels of CD22. Data are presented as mean±SEM (PBMCs from n=3 healthy donors). *p<0.05, **p<0.01. ANOVA, analysis of variance; B-ALL, B-cell acute lymphoblastic leukemia; CAR, CD22-chimeric antigen receptor; E:T, effector:target ratio; FACS, fluorescence-activated cell sorting; MFI, median fluorescence intensity; FSC, forward scatter; FSC-A, forward scatter-area; FSC-H, forward scatter-high; PBMCs, peripheral blood mononuclear cells; SSC, side scatter.
Membrane-distal CD22 epitope-directed CAR T cells efficiently eliminate B-ALL blasts in vivo in clinically relevant patient-derived xenografts
We next assessed the activity of our hCD22.7-CAR in vivo using both CD22high (ALL#1 and ALL#2) and CD22low (ALL#10) PDX B-ALL samples (figure 4, online supplementary table 1). NSG mice were intra-BM transplanted with 0.5–1.5×106 primary CD22+ B-ALL blasts, followed by infusion of 4×106 CD22-CAR/mock-IC T cells 2 weeks later. Leukemic engraftment and T cell persistence were monitored by FACS biweekly in PB, and analyzed in BM and spleen at sacrifice, when any animal developed signs of overt disease (figure 4A,B). Of note, the kinetics of leukemia reconstitution (aggressiveness) detectable between week 4 and 20 post-transplant was unique for each PDX, and the engraftment of both CD22high and CD22low PDXs tended progressively to increase over time in mock-IC T cell-treated mice (figure 4C). Importantly, however, hCD22.7-CAR T cells eradicated (ALL#1 and ALL#10) or largely controlled (ALL#2) the growth of the both CD22high and CD22low B-ALL cells (figure 4C,D). T cell persistence adapted to the reconstitution kinetics of each PDX, and was detected in all three PDXs, even 26 weeks after CAR T cell infusion (range, week 6 (n=15) to week 26 (n=5): mock-IC group 2.38%±0.65%–0.34%±0.34%; CAR T cell group 2.79%±1.13%–0.14%±0.09%, Figure 4E and online supplementary figure 4a-b). Disease control was further confirmed at sacrifice in ipsilateral (injected tibia) and contralateral (both femurs and non-injected tibia) BM, PB and spleen through FACS quantification of tumor burden (figure 4F and online supplementary figure 4c), lack of splenomegaly (figure 4G), and signs of hematopoietic displacement (figure 4H). Importantly, persistent leukemic cells at sacrifice maintained expression of CD22, likely ruling out CAR pressure-mediated antigen loss (figure 4I).

CAR T cells directed against a distal membrane CD22 epitope efficiently eliminate differentially aggressive PDX B-ALL blasts in vivo. (A) Experimental design of the in vivo experiments. (B) Gating strategy used to analyze FACS data from the in vivo experiments. Leukemic cells were identified as hCD45+hHLA-ABC+hCD3–hCD19+hCD10+. CD22 was excluded from the identification to avoid confounding analysis due to potential antigen loss. CAR T cells were identified based on CD3 and GFP expression. Doublets were removed from the analysis. (C) Leukemic burden measured in PB over a 26 week follow-up period (n=4–5 mice/group; n=3 PDXs, CD22high (ALL#1 and ALL#2) and CD22low (ALL#10)). (D) Disease-free survival curves of mice transplanted with the three patient derived xenograft (PDXs). A mouse was considered ‘with disease’ when presenting >0.1% blasts in PB. (E) T cell persistence measured in PB of mice transplanted with the three PDXs. (F) Leukemic burden at sacrifice in PB, ipsilateral and contralateral BM and spleen (n=9 mice from the ALL#2 PDX). Each mouse is identified with the same number across the plots. Macroscopic images of these spleens are also shown. (G) Spleen weight (total animals, n=14) from mock-IC and CD22-CAR T cell-treated mice at sacrifice. (H) PB RBC counts in mock-IC and CD22-CAR T cell-treated mice at sacrifice (n=14). (I) CD22 MFI of persistent primary B-ALL in mock-IC-treated and CD22-CAR T cell-treated mice at sacrifice (ALL#2 mice, n=4–5) and representative FACS plots of contralateral BM samples showing the levels of CD22 of a mock-IC and a CD22-CAR T cell-treated mouse within the target cell population. Plot shows mean±SEM. *p<0.05, **p<0.01, ***p<0.001. B-ALL, B-cell acute lymphoblastic leukemia; BM, bone marrow; CAR, CD22-chimeric antigen receptor; D, day; FACS, fluorescence-activated cell sorting; PB, peripheral blood; it, intratibia; iv, intravenous; MFI, median fluorescence intensity; mock-IC, mock intracellular; RBC, red blood cell; W, week.
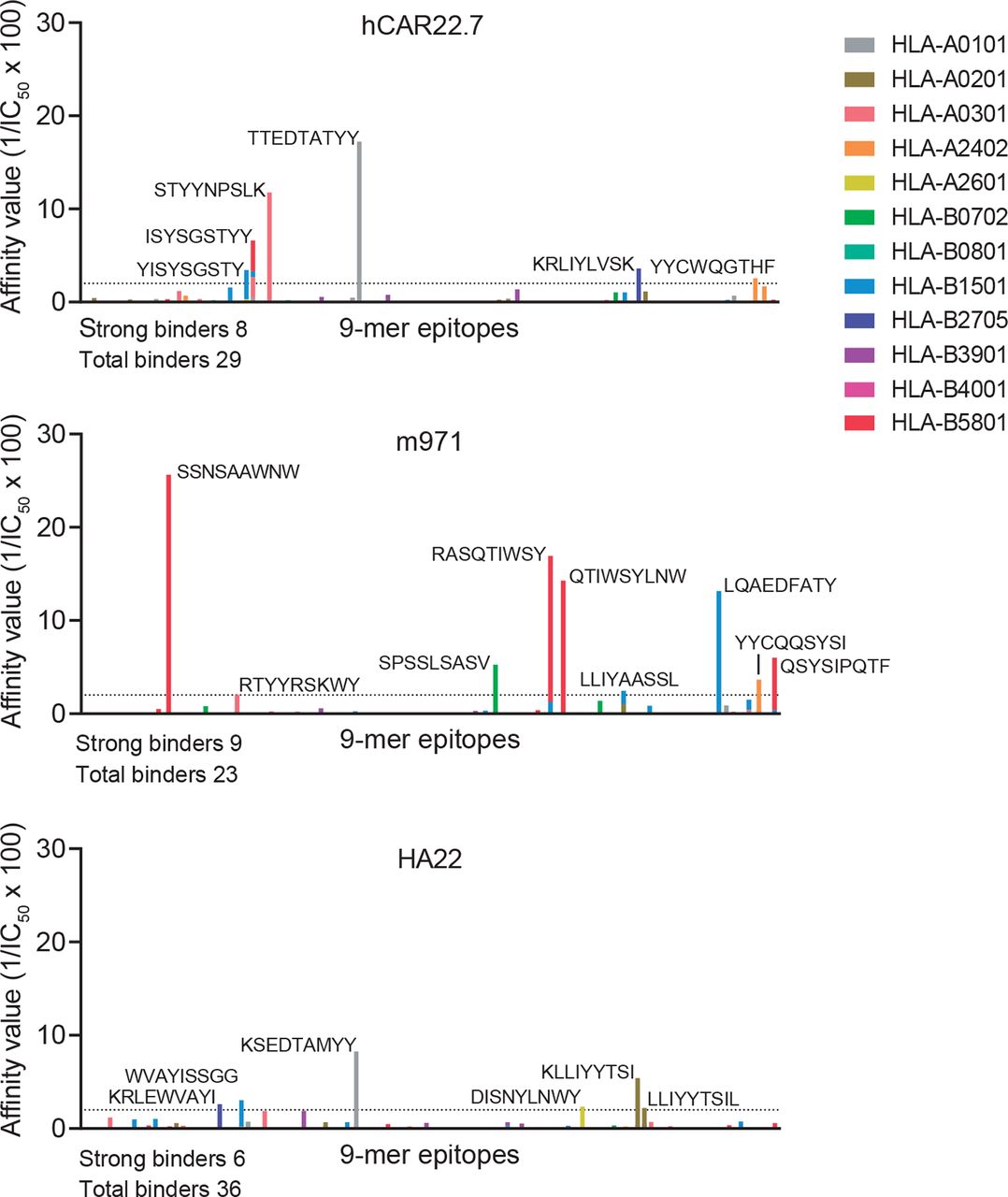
Finally, any engineered scFv of either murine or human origin expressed in autologous T cells may be immunogenic if presented on MHC-I, because peptides derived from it may be recognized as ‘non-self’ antigens by the adaptive immune system.39 We have thus compared the immunogenicity capacity of our hCD22.7-scFv side-by-side with that of m97115 and HA2220 clones using the in silico tool NetMHC V.4.0 which predicts the affinity of peptides to bind to the most representative HLA supertypes. As shown in figure 5, we found very similar numbers of total (29, 23 and 26) and strong (8, 9 and 6) binding 9-mer peptides for hCD22.7, m971 and HA22, respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immunogenic prediction of hCD22.7-scFv. The immunogenic potential of hCD22.7, m971 and HA22 CD22-scFvs was compared by in silico prediction analysis. CD22-scFvs were broken in 9-mer peptides, length for which human leukocyte antigen (HLA) molecules have a strong preference. Peptides are considered to bind MHC-I when a IC50 is <500 nM, being considered strong binders when IC50 <50 nM (affinity value >2, dotted line) and amino acid sequence is shown in the figure. Only the most representative HLA supertypes were analyzed. The predicted epitope-binding affinity was calculated as 1/IC50 100. Predicted epitopes are distributed along the x-axis according to their distribution in the protein. IC50, half-maximum inhibitory concentration; scFv, single-chain variable fragment.
Discussion/Conclusions
CD19-CAR T cell therapy has shown indisputable potential in R/R B-ALL.9 10 Unfortunately, however, 40%–60% of patients ultimately relapse because of poor CAR T cell persistence or the emergence of CD19– B-ALL clones.11 12 Very few therapeutic options exist for CD19– B-ALL relapse,40 41 and novel strategies, including the development of new target antigens, are being explored to enhance CAR T cell function and persistence.13 42 CD22 is a pan B-cell antigen,14 and is an attractive alternative to CD19 for CAR T cell therapy because it will target CD19– relapses, which usually retain CD22 expression.
In this line, several CD22-CARs have been reported to induce clinical remission in 60%–70% of patients with R/R B-ALL either naive or resistant to CD19-CAR T cells.15 20–22 But, phase I clinical studies based on different CD22 scFvs (m971 vs YK-CD22BB-002) have reported preliminary results concerning CD22 loss and immune escape on CD22-CAR T cell treatment.15 21 A previous comprehensive comparison of several anti-CD22 moAbs indicated that epitope selection is crucial for the antileukemic activity of the CD22-CAR, and that the m971 scFv, which targets the most membrane-proximal Ig extracellular domains of CD22 (5 to 7), seems more potent than BL22 and HA22 scFvs, which target the proximal Ig domain 3.20 In this study, we report the generation and preclinical development of a new CD22-CAR that contributes to the limited state-of-the-art arsenal of CD22-CARs. Three CD22 reactive hybridomas were generated using conventional moAb generation techniques, rendering three very specific IgG1 isotype anti-CD22 moAbs, finally selecting the hCD22.7 clone for the construction of the CAR. To the best of our knowledge, this ab initio generated hCD22.7 scFv is the first used for the development of a CD22-CAR recognizing the most membrane-distal Ig extracellular domain 1 of CD22. Additionally, we provide an uncommon comprehensive characterization including the molecular docking, epitope mapping, binding affinity, and immunogenicity of the hCD22.7 scFv.
Previous studies have addressed the impact of antigen density on CD22-CAR T cell efficacy using a higher-affinity version of the m971 scFv, and reported a positive correlation between CD22 expression and the functionality of CD22-CAR T cells, both in vitro and in vivo, using cell lines and one patient derived xenograft (PDX).22 Here, the expression level of CD22 was used to classify primary B-ALL samples as CD22high or CD22low, and we show that although our high-affinity hCD22.7-CAR efficiently and consistently targeted CD22+ cells, it displayed a differential killing kinetics depending on the expression level of CD22. While a sustained cell elimination of CD22high cells was observed over a 48 hours period, a shorter or delayed but still efficient cytotoxic window was observed for CD22low cells. It is also plausible that CD22 adopts different conformational epitope exposures43 affecting the performance of the CAR T cells in the different samples. Of note, a robust production of proinflammatory cytokines was observed for all B-ALL primary samples, regardless the expression levels of CD22, confirming an efficient CD22 recognition and killing of B-ALL primary cells by our hCD22.7-CAR T cells.
Our membrane distal epitope hCD22.7-CAR T cells performed competently in controlling in vivo several B-ALL PDXs with varied aggressiveness for a long period, which was coupled to long-term T cell persistence. In fact, hCD22.7-CAR T cells were capable of eradicating long-term disease in several PDXs, with persistence of T cells even after 26 weeks. In the PDX ALL#2, although the leukemia burden was not fully eradicated, it was significantly controlled. The not complete eradication of this PDX may reflect a more aggressive molecular subtype, a superior intrinsic refractoriness due to resistance generated through multiple lines of previous treatments, a faster/deeper graft of this particular PDX, a worse pharmacodynamics of CAR T cells in this particular case perhaps due to peripheral filtration, and so on. Of note, we found no apparent signs of CD22 antigen loss by the few surviving/resistant B-ALL cells in vivo. Antigen loss represents one non-exclusive potential mechanism of immune escape and largely relies on tumor-specific cell-autonomous properties, differentiation stage in which leukemic cells are stalled, and the complexity of immune cellular and soluble interactions, difficult to reconstruct in xenograft models. In addition, it cannot be ruled out that residual CAR-resistant CD22+ leukemic cells have simply not been encountered by CD22-CAR T cells. Only a controlled and homogeneous phase I clinical trial will reliably inform about a potential target antigen loss and immune scape. Technically, our experimental design used a rigorous ‘mock’ control where all the structural, cytolytic and costimulatory motifs are expressed in the effector T cells, but without the extracellular anti-CD22 scFv region. This intracellular mock further validates the specificity and sensitivity of our hCD22.7-CAR.
Our hCD22.7-derived membrane distal-targeting CAR has not been functionally compared side-by-side with other membrane proximal-targeting CD22-scFvs used already in clinical trials,15 21 with which Fry et al published discrete complete remission (CR) rates of 57% with a median remission duration of 6 moths in patients with R/R B-ALL treated with m971-CD22-CAR T cells15 and in a more recent study they reported CR of 70% with a median relapse-free survival of 6 months.23 However, the hCD22.7-CAR performs efficiently in vitro and in vivo using clinically relevant patient samples of B-ALL with different aggressiveness. This coupled to the long-term persistence of hCD22.7-CAR T cells, the lack of antigenic loss in vivo in the few CAR-resistant B-ALL cells, its high affinity, and its immunogenicity similar to m971 and HA22 CD22-scFvs provides support for its clinical validation as either single or tandem CD22–CD19-CAR for both naive and anti-CD19-resistant patients with B-ALL.
Supplemental material
Acknowledgments
We thank CERCA/Generalitat de Catalunya and Fundació Josep Carreras‐Obra Social la Caixa for their institutional support.
References
Footnotes
Contributors TVH conceived the study, designed and performed experiments, analyzed data, and wrote the manuscript. SRZ designed and performed experiments. HRH, FGA, PP, MLB, OM, and DSM performed experiments. NFF generated the structural model. PE generated the CD22 hybridoma and performed experiments. JLF and PB provided primary samples. CB supported the study technically and financially. PM conceived the study, designed experiments, wrote the manuscript, and financially supported the study.
Funding This work was supported by the European Research Council (CoG-2014-646903, PoC-2018-811220), the Spanish Ministry of Economy and Competitiveness (MINECO-SAF2016-80481-R), the Fundación Uno entre Cienmil, the Obra Social La Caixa (LCF/PR/HR19/52160011), the Spanish Cancer Association and Leo Messi Foundation to PM. CB was supported by the ISCIII/FEDER (PI17/01028). TVH and SRZ were supported by Marie Sklodowska Curie Fellowships (792923 and 795833, respectively). DSM was supported by a Sara Borrell fellowship from the ISCIII. MLB was supported by a FPI scholarship from the MINECO. PM is an investigator of the Spanish Cell Therapy Network (TERCEL).
Competing interests PM is co-founder of OneChain Immunotherapeutics (OCI), a IJC spin-off company focused on the development of CAR T-cell therapies. The patent of the membrane-distal CD22 scFv clone has been licenced to OCI.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data sharing not applicable as no datasets generated and/or analyzed for this study. Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.