Article Text

Abstract
Background CMP-001 is a novel Toll-like receptor-9 agonist that consists of an unmethylated CpG-A motif-rich G10 oligodeoxynucleotide (ODN) encapsulated in virus-like particles. In situ vaccination of CMP-001 is believed to activate local tumor-associated plasmacytoid dendritic cells (pDCs) leading to type I interferon secretion and tumor antigen presentation to T cells and systemic antitumor T cell responses. This study is designed to investigate if CMP-001 would enhance head and neck squamous cell carcinoma (HNSCC) tumor response to anti-programmed cell death protein-1 (anti-PD-1) therapy in a human papilloma virus-positive (HPV+) tumor mouse model.
Methods Immune cell activation in response to CMP-001±anti-Qβ was performed using co-cultures of peripheral blood mononuclear cells and HPV+/HPV- HNSCC cells and then analyzed by flow cytometry. In situ vaccination with CMP-001 alone and in combination with anti-PD-1 was investigated in C57BL/6 mice-bearing mEERL HNSCC tumors and analyzed for anti-Qβ development, antitumor response, survival and immune cell recruitment. The role of antitumor immune response due to CMP-001+anti-PD-1 treatment was investigated by the depletion of natural killer (NK), CD4+ T, and CD8+ T cells.
Results Results showed that the activity of CMP-001 on immune cell (pDCs, monocytes, CD4+/CD8+ T cells and NK cells) activation depends on the presence of anti-Qβ. A 2-week ‘priming’ period after subcutaneous administration of CMP-001 was required for robust anti-Qβ development in mice. In situ vaccination of CMP-001 was superior to unencapsulated G10 CpG-A ODN at suppressing both injected and uninjected (distant) tumors. In situ vaccination of CMP-001 in combination with anti-PD-1 therapy induced durable tumor regression at injected and distant tumors and significantly prolonged mouse survival compared with anti-PD-1 therapy alone. The antitumor effect of CMP-001+anti-PD-1 was accompanied by increased interferon gamma (IFNγ)+ CD4+/CD8+ T cells compared with control-treated mice. The therapeutic and abscopal effect of CMP-001+ anti-PD-1 therapy was completely abrogated by CD8+ T cell depletion.
Conclusions These results demonstrate that in situ vaccination with CMP-001 can induce both local and abscopal antitumor immune responses. Additionally, the antitumor efficacy of CMP-001 combined with α-PD-1 therapy warrants further study as a novel immunotherapeutic strategy for the treatment of HNSCC.
- adjuvants
- immunologic
- head and neck neoplasms
- dendritic cells
- vaccination
- immunotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Immunotherapy with antibodies targeting programmed cell death protein-1 (PD-1) has shown great promise for head and neck squamous cell carcinoma (HNSCC).1–3 Anti-PD-1 therapy has only modest response rates (<20%); however, these responses are remarkably durable.1–3 Nevertheless, the problem remains that only a minority of patients with HNSCC derive benefit from anti-PD-1-based therapy. As a result, there remains a need for the development of additional novel immunotherapeutic concepts for HNSCC treatment that will improve the rate of durable responses beyond what is observed with anti-PD-1 agents.
Anti-PD-1 agents are believed to be ineffective at triggering antitumor immune responses in poorly immunogenic tumors which characterize the vast majority of tumors.4 The number of tumor-infiltrating lymphocytes (TILs) plays a significant role in determining whether a tumor is highly immunogenic (hot) or poorly immunogenic (cold). However, the proliferation and the activity of TILs are dependent on the maturation and activation status of dendritic cells (DCs).5 In many cases, pre-existing DC activity is absent or limited in poorly immunogenic tumors, which impedes on the ability of anti-PD-1 agents to trigger an antitumor immune response.6
DCs can be activated through Toll-like receptor (TLR) stimulation,7 8 which is currently being investigated as an alternate and adjuvant to standard cancer therapies.9 TLRs are transmembrane proteins that are expressed on the cell surface and on endosomes.10 TLRs recognize structurally conserved molecules derived from microbes and are involved in the development of the adaptive immune response.10 Of the 10 TLRs identified in humans (TLR1-10), TLRs 7–9 are located in endosomes and recognize virus-associated nucleic acids and signals through the myeloid differentiation factor 88 (MyD88)-dependent pathway which ultimately leads to nuclear factor kappa B (NF-κB) activation.11 TLR9 in particular is highly expressed in the endosomes of plasmacytoid dendritic cells (pDCs) and B cells and are activated by unmethylated CpGs in bacterial and viral DNA which triggers the trafficking of TLR9 from the endoplasmic reticulum through the Golgi to early endosomes.11 12 TLR9 activation in early endosomes leads to the phosphorylation of IRF7, interferon gamma (IFNα) secretion, and activation of an innate immune response.13 In late endosomes, TLR9 triggering preferentially activates NF-κB, resulting in the secretion of interluekin-6 (IL-6) and tumor necrosis factor-6 (TNFα), and thus an adaptive immune response.13
TLR9 agonists are believed to trigger monocyte maturation into functional DCs and directly induce activation and maturation of pDCs, leading to the secretion of IFNα (and other cytokines), activation of natural killer (NK) cells and expansion of T cell populations especially CD8+ T cells.14 TLR9 agonists also promote B cell differentiation, proliferation, immunoglobulin class switching and antibody production. TLR9 agonists such as CMP-001, IMO-2125, SD101, ODN-1826 and MGN1703 are synthetic CpG oligodeoxynucleotides (ODNs) that are being tested for cancer treatment.15–20 CMP-001 differs from the other existing TLR9 agonists in that the ODN is a CpG-A which stimulates high amounts of type 1 IFN from pDCs compared with CpG-B and CpG-C.21 22 The CpG-A ODN is also encapsulated in virus-like particles (VLPs) composed of the Qβ bacteriophage capsid protein.22 This encapsulation protects the CpGs from premature degradation before APC uptake and is of an appropriate size (~30 nm) allowing for their free draining into lymph nodes (LNs) and triggers an antitumor immune response in the presence of anti-Qβ.23 24
Clinical trials are currently being conducted with CMP-001 in combination with PD-1 blockade in multiple tumor types including melanoma, colon, HNSCC, and lymphoma. Encouraging preliminary data have been reported in patients with unresectable or metastatic melanoma previously treated with anti-PD-1 therapy with CMP-001 combined with anti-PD-1 (pembrolizumab) (NCT02680184), and in patients with resectable metastatic melanoma treatment naïve to PD-1 blockade with CMP-001 in combination with nivolumab (NCT03618641).25 26 The goal of the current preclinical studies is to investigate if in situ vaccination with CMP-001 will trigger both local and systemic antitumor immune responses in HNSCC tumors. Here, we show that in situ vaccination with CMP-001 can induce both local and abscopal antitumor immune responses in the presence of anti-Qβ and that CMP-001 can significantly enhance tumor response to anti-PD-1 therapy.
Materials and methods
Cell lines and reagents
The UM-SCC47 cell line was obtained from Millipore Sigma. The SQ20B cell line was a gift from Dr Anjali Gupta (Department of Radiation Oncology, The University of Iowa, Iowa, USA). The murine oropharyngeal epithelial cell line stably transformed with HPV E6 and E7 together with hRas and luciferase (mEERL) was a gift from Dr Paola D. Vermeer (Department of Surgery, University of South Dakota Sanford School of Medicine, South Dakota, USA). SQ20B and UM-SCC47 cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS) and 0.1% gentamicin, and mEERL was cultured in DMEM supplemented with 40.5% 1:1 DMEM/Hams F12, 10% FBS, 0.1% gentamicin, 0.005% hydrocortisone, 0.05% transferrin, 0.05% insulin, 0.0014% tri-iodothyronine and 0.005% epidermal growth factor. All cell lines were authenticated by short-tandem repeat profiling and used over a course of no more than 3 months after resuscitation of frozen aliquots. Cells were cultured in a humidified incubator at 37°C and 5% CO2. CMP-001, empty VLPs and recombinant anti-Qβ Ig were provided by Checkmate Pharmaceuticals. Non-methylated CpG-A ODN (G10; 5′-GGG GGG GGG GGA CGA TCG TCG GGG GGG GGG-3′) and its manufacture process were described in previous work.15 Anti-human PD-1 IgG1 antibody (clone: J116) was purchased from BioXCell.
In vitro immune cell activation
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donor blood obtained from the DeGowin Blood Center (University of Iowa Hospitals and Clinics) by Ficoll density gradient centrifugation. PBMCs were treated with CMP-001 (5 µg/mL) with or without recombinant anti-Qβ IgG (5 µg/mL) for 24 hours before analysis of immune cells by flow cytometry. PBMCs were stained with anti-CD45, anti-CD3, anti-CD19, anti-CD11c, anti-BDCA-4, anti-CD123, anti-CD16, anti-CD14, anti-human HLA-DR and anti-CD40 conjugated to different fluorochromes. pDCs were defined as BDCA-4+ CD123+ CD45+ CD3- CD19- CD11c- immune cells, and monocytes were defined as CD45+ CD3- CD19- CD11c+ CD14+ immune cells. CD40+ pDCs and CD40+ monocytes were considered activated. For co-culture systems, human papilloma virus-negative (HPV-) SQ20B or human papilloma virus-positive (HPV+) UM-SCC47 cell lines were 1:1 cultured with human PBMCs in 96-well plates, treated with CMP-001 (5 µg/mL) and recombinant anti-Qβ IgG (5 µg/mL) and then PBMCs stained with anti-CD45, anti-CD3, anti-CD19, anti-CD8, anti-CD4, anti-CD56, anti-CD69 and anti-CD107a. Activated T cells were defined as CD3+ CD4/CD8CD69+ and activated NK cells were defined as CD3- CD19- CD56+ CD69+. T-distributed Stochastic Neighbor Embedding (T-SNE) map was generated using flow cytometry data by FlowJo V.10.4. Briefly, samples were concatenated and a subset of cell population (20 000 cells) was analyzed at single cell level with perplexity of 20 and iteration number of 1000. All values from flow cytometric experiments were quantified based on percentage positive cells and fold change plotted compared with control. IFNγ and TNFα concentration levels in the media from the co-culture assays were quantified using Human Quantikine ELISA kits (R&D Systems).
Tumor cell implantation
Female C57BL/6J mice (6–8 weeks) were purchased from The Jackson Laboratory. Mice were housed in a pathogen-free barrier room in the Animal Care Facility at the University of Iowa and handled using aseptic procedures. All procedures were approved by the IACUC committee of the University of Iowa and conformed to the guidelines established by the National Institutes of Health. mEERL cells (1×106 cells/mouse) were inoculated into left and right flank of C57BL/6 mice by subcutaneous injection of 0.1 mL aliquots of saline containing cancer cells. For tumor implantation, mice were anesthetized with ketamine (80 mg/kg)/xylazine (10 mg/kg) purchased from the inpatient pharmacy at the University of Iowa Hospital and Clinics.
In vivo drug administration
For the anti-Qβ development studies, mice (n=10/treatment group) were subcutaneously injected with empty VLP or CMP-001 (100 µg/mouse) before tumor challenge as a ‘priming’ step. Blood samples from a subset of mice (n=4–5) were collected 1 week (Day −7) and 2 weeks (Day 0) after priming and analyzed for immunoglobulin (Ig) anti-Qβ concentration by ELISA. ‘Non-primed’ mice were used as a control. After tumor formation (3–5 mm in any dimension), empty VLP or CMP-001 was administered to the left tumor only intratumorally on Days 14, 17 and 20. In a separate experiment, CMP-001, empty VLP, G10 CpG ODN (G10) or succinate buffer was administered intratumorally to the tumor on the left flank of mice (n=10/treatment group) on Days 14, 17 and 20 after tumor inoculation. All mice were primed 2 weeks before tumor inoculation with their respective assigned treatments. For the combined CMP-001+ anti-PD1 experiments, mEERL tumor-bearing C57Bl/6 mice (n=9–12/treatment group) were treated with CMP-001 with or without anti-mouse PD-1 (rat IgG2a, clone RMP1-14). IgG isotype control in succinate buffer was used as a control. CMP-001 and succinate were administered intratumorally into the left tumor on Days 11, 15 and 19, and IgG and anti-PD1 were administered intraperitoneally on Days 11, 15, 19, 24, 28, and 32 after mEERL tumor inoculation. All mice receiving CMP-001 were primed 2 weeks before tumor inoculation. For the immune cell depletion experiments, murine anti-CD4 mAb (clone GK1.5) and anti-CD8 mAb (clone 53–6.7) were purchased from BioXcell, and polyclonal anti–asialo GM1 antibody was purchased from Thermo Fisher Scientific. C57BL/6 mice (n=6–8 mice/group) bearing mEERL tumors were administered CMP-001+anti-PD-1 antibody or IgG isotype control in succinate buffer as described above with or without anti-CD4 (100 µg), anti-CD8 (300 µg) and anti–asialo GM1 (50 µg). The immune cell depleting antibodies were given 3 days and 1 day before tumor inoculation and twice per week after tumor inoculation. T and NK cell depletion in vivo was validated using flow cytometry. For all experiments, tumor volume for the left (injected) and right (distant (uninjected)) tumors in the treated mice was measured three to four times per week using Vernier calipers, and overall survival was recorded when mice reached euthanasia criteria.
Immune cell infiltration and circulating cytokines
Lingual lymph nodes (LNs) were harvested 1–2 days after the last drug treatment. Tissues were dissociated to produce single cell suspensions (gentleMACS Dissociator, Miltenyi Biotec). Cells were incubated with ACK buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.3) for 2 min to remove red blood cells and then washed by fluorescence-activated cell sorting (FACS) buffer. Cells were stained with Zombie Aqua Fixable Viability dye (Biolegend) and cocktails of antibodies (CD45.2, CD3e, CD4, CD8α, CD11b, CD11c, CD19, CD335, F4/80, Ly6G, Ly6C, and MHC II, Biolegend) at 4°C for 30 min protected from light. For intracellular cytokine staining, Fixation/Permeabilization Solution kits (eBioscience) were applied. Cells were stained with antibodies against FoxP3 and IFNγ (Biolegend). HPV+ CD8+ T cells in LNs were detected by HPV E7-specific iTAg tetramer staining (PE-H-2Db HPV 16 E7 (RAHYNIVTF), MBL International Corporation). After staining, CountBright Absolute Counting Beads (Thermo Fisher) were added to the samples to obtain absolute immune cell numbers. Cells were fixed by 2% paraformaldehyde (450 µL/tube) and were analyzed by flow cytometry using an LSR Violet Flow Cytometer (BD Biosciences). The number of tumor-infiltrating immune cells was normalized by tumor volume and tumor weight. The concentrations of proinflammatory analyte cytokines in the mouse sera were determined using a mouse Bio-Plex 23 panel assay, as per the manufacturer’s instructions (Bio-Rad), and quantified using a Bio-Plex array reader. Bio-Plex Manager software was used to calculate analyte concentrations.
The Cancer Genome Atlas analysis
A dataset of gene expression of 520 HNSCC patients (TCGA_HNSC_exp_HiSeqV2-2015-02-24) along with the corresponding clinical outcomes were downloaded from The Cancer Genome Atlas (TCGA) using Xena Functional Genomics Explorer (University of California – Santa Cruz). Patients were divided into two groups according to their TLR9 gene expression levels and labeled as ‘high’ (n=284) and ‘low’ (n=236) TLR9 gene expression. The two gene expression groups were analyzed for differences in overall survival (using Kaplan-Meier curves) and levels of activated CD4+ T cells and CD8+ T cells. Expression of immune cell population in tumors was estimated using CIBERSORT algorithm based on gene expression of 22 types of flow-purified immune cell population.
Statistics
Statistical analysis was carried out using GraphPad Prism V.8 for Windows (GraphPad Software, San Diego, California, USA). One-way analysis of variance (ANOVA) with Tukey post-tests was used to compare the difference between at least three treatment groups. Two-way ANOVA was used to compare differences between cell lines and treatment groups. Linear regression models were used to estimate the group-specific change in tumor growth curves. Kaplan-Meier survival curves were generated to illustrate the different survival rates over time. Differences in survival were determined by log-rank (Mantel-Cox) test. Fisher’s exact test was used to analyze any associations of TLR9 expression with patient characteristics. Statistical significance was defined as p<0.05.
Results
Anti-Qβ is required for CMP-001-induced immune cell activation in vitro
To determine if pDCs could be activated by CMP-001, human PBMCs were incubated with CMP-001±anti-Qβ to analyze pDC activation by flow cytometry. CMP-001 significantly activated pDCs and monocytes (identified by CD40-positivity) but only in the presence of recombinant anti-Qβ IgG (figure 1A.B). To determine if DC activation would drive T and NK cell activation in the presence of CMP-001, co-cultures of HPV+ (UM-SCC47) or HPV- (SQ20B) HNSCC cell lines and PBMCs were treated with CMP-001 with or without anti-Qβ. Flow cytometric and t-SNE analyses revealed that CMP-001+anti-Qβ significantly increased CD69-positivity in select immune subsets compared with control, as illustrated in representative tSNE plots in figure 1C,D, and compared with CMP-001 or anti-Qβ alone including CD4+ (figure 1E) and CD8+ T cells (figure 1F) and NK cells (figure 1G). These results were only observed in the HPV+ UM-SCC47/PBMC co-cultures, although there was a trend of increased immune cell activation in the HPV- SQ20B/PBMC co-cultures (figure 1E–G). Additionally, CMP-001+anti-Qβ was unable to increase CD4+ or CD8+ T cell activation in isolated T cell/cancer cell co-cultures (data not shown), suggesting that the presence of DCs is important for T cell activation. However, there was a significant increase in NK cell activation compared with the other treatment groups when isolated NK cells were co-cultured with the UM-SCC47 cell line, although this increase was not as robust as with PBMCs (approximately threefold (isolated NK cells) vs eightfold (PBMCs), online supplemental figure 1). Secreted levels of IFNγ (figure 1H) and TNFα (figure 1I) in cell culture media were also increased by CMP-001+anti-Qβ compared with the other treatment groups in the HPV+ UM-SCC47/PBMCs co-cultures only. Altogether, these results suggest that CMP-001 in the presence of anti-Qβ can directly induce DC activation, leading to T cell activation. Additionally, HPV+ cancer cells may be more sensitive to CMP-001-based therapy.
Supplemental material

Activity of CMP-001 on immune cell infiltration depends on the presence of anti-Qβ. (A, B) Peripheral blood mononuclear cells (PBMCs) were treated with CMP-001 (5 µg/mL) and/or anti-Qβ (5 µg/mL) for 24 hours. Activation of plasmacytoid dendritic cells (pDCs) (CD45+ CD3- CD19- CD11c- BDCA-4+) (A) and monocytes (CD45+ CD3- CD19- CD11c+ CD14+) (B) was monitored by flow cytometry. (C–I) SQ20B (human papilloma virus-negative (HPV-)) and SCC47 (human papilloma virus-positive (HPV+)) cells were co-cultured with PBMCs, treated with CMP-001±anti-Qβ as previously described, then flow cytometry and T-distributed Stochastic Neighbor Embedding (t-SNE) data analysis (C,D) was used to identify activated CD4+ (E), CD8+ (F), and natural killer (NK) cells (G), and interferon gamma (IFNγ) (H) and tumor necrosis factor α (TNFα) (I) concentrations in cell culture media analyzed by ELISA. Succinate buffer was used as a control (CON). Representative t-SNE plots are shown for CON (C) and CMP-001+anti-Qβ (D). Bar graphs shown represent the mean of n=3 experiments. Error bars represent SD from the mean. *p<0.05 versus CON; **p<0.05 versus CMP-001; #p<0.05 versus UM-SCC47.
Anti-Qβ development is important for favorable CMP-001-induced tumor responses
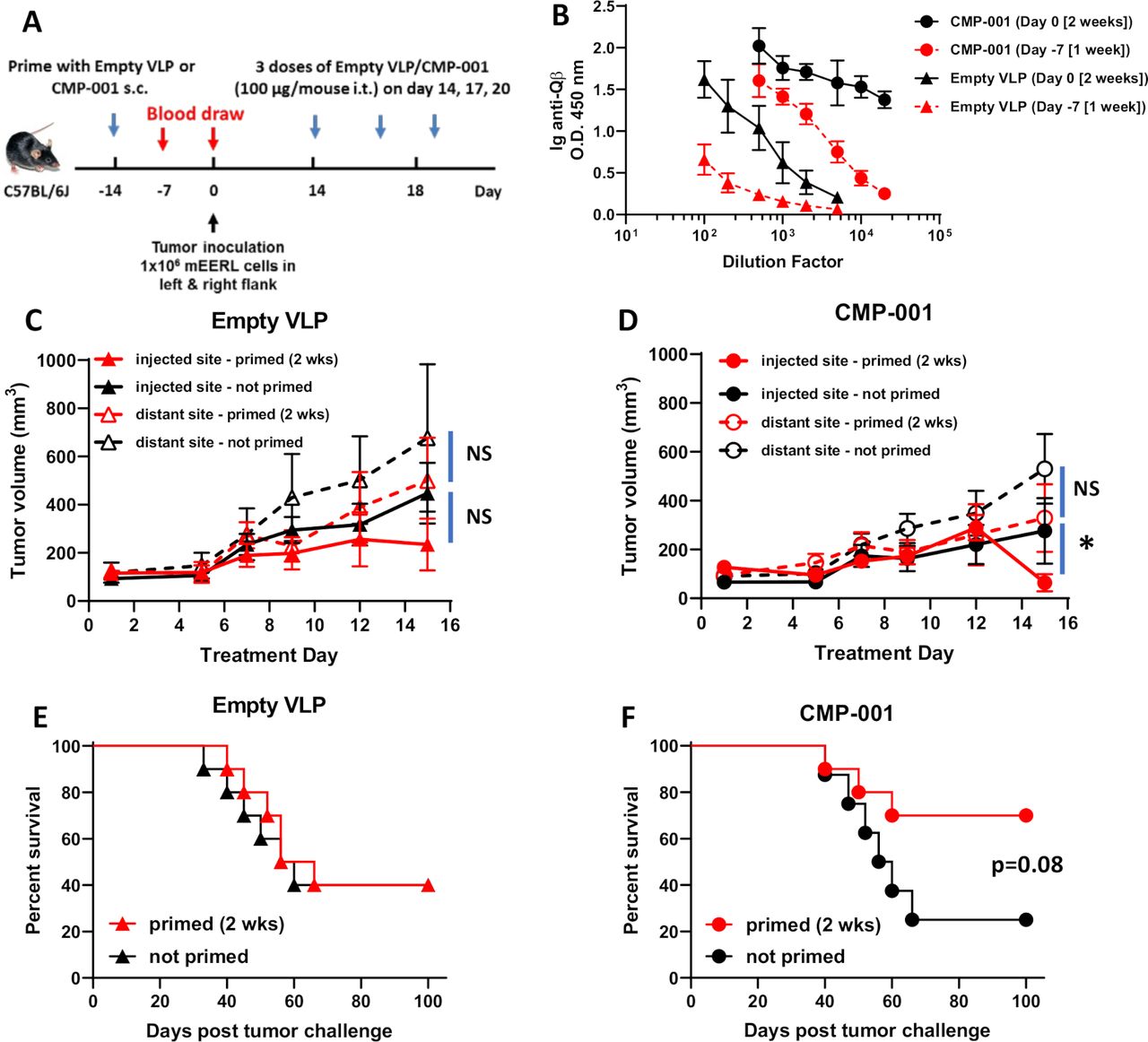
Given that anti-Qβ antibody was necessary for CMP-001-induced immune cell activation in vitro (figure 1), we investigated the time required for anti-Qβ antibody development after administration of CMP-001 or empty VLP subcutaneously to C57Bl/6 mice (figure 2A). A more robust anti-Qβ response was observed after 2 weeks for both CMP-001 and empty VLPs compared with 1-week post-VLP administration (figure 2B). Anti-Qβ levels were also generally higher after priming with CMP-001 compared with empty VLPs regardless of time, suggesting that CMP-001-induced TLR9 signaling in B cells may be further promoting antibody production (figure 2B). CMP-001 or empty VLPs were then administered subcutaneously to mice 2 weeks as a ‘prime’ step before inoculation with mEERL tumor cells on both left and right flanks of each mouse (figure 2A). After tumor formation, CMP-001 or empty VLPs were administered to each mouse intratumorally to the left flank tumor every 3 days. ‘Non-primed’ mice were used as controls. There were no differences in injected or distant tumor growth between primed and non-primed mice administered empty VLPs (figure 2C) or CMP-001 (figure 2D) during the initial 2-week treatment period. Although on Day 15, the injected tumors in primed CMP-001 mice began to regress compared with non-primed CMP-001 mice (figure 2D). There was also no difference in survival between ‘primed’ and ‘non-primed’ mice when treated with empty VLPs (figure 2E), but a clear difference in survival was observed in CMP-001-treated mice between primed and non-primed mice, although this difference did not quite meet significance (p=0.08, figure 2F). These results suggest that pre-existing anti-Qβ may boost antitumor activity of CMP-001.

A 2-week ‘priming’ is required for robust anti-Qβ development and favorable survival outcomes. (A) The treatment schema for the priming of C57BL/6 mice (n=10/treatment group) with empty virus-like particle (VLP) or CMP-001 (100 µg/mouse) subcutaneously (s.c.) 2 weeks or 1 week before tumor challenge with mEERL cells on both left and right flanks. (B) Blood samples from a subset of mice (n=4–5) were collected 1 week (Day −7) and 2 weeks (Day 0) after priming and analyzed for immunoglobulin (Ig) anti-Qβ concentration. After tumor formation, empty VLP or CMP-001 was then administered to the left tumor only intratumorally (i.t.) every 3 days. Tumor volume (C, D) for the left (injected) and right (distant (uninjected)) tumors in primed and unprimed empty VLP (C) and CMP-001 (D) treated mice was measured three to four times per week. Tumor growth curves shown were terminated when a mouse in any treatment group reached euthanasia criteria. Overall survival was analyzed by constructing Kaplan-Meier plots for empty VLP (E) and CMP-001 (F) treated mice. Error bars represent SD from the mean. *p<0.05. NS, non-significant. Note: Treatment Day 1 in tumor growth graphs (C, D) represent Day 14 in treatment schema in (A).
Antitumor effects of CMP-001 are superior to unencapsulated G10 CpG ODN
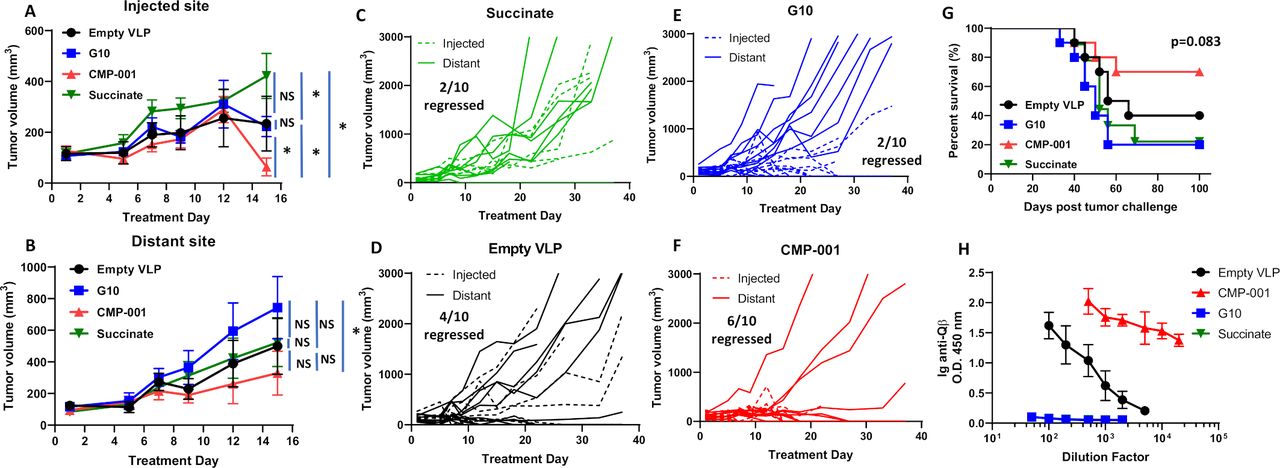
To determine if the antitumor effect of CMP-001 was superior to the unencapsulated G10 CpG ODN (G10), C57BL/6 mice bearing mEERL tumors on both flanks were treated intratumorally with succinate as a control, G10, empty VLP and CMP-001 into the left tumor only as described in figure 2A. All mice were primed 2 weeks before tumor inoculation with their respective assigned treatments. G10 and CMP-001 significantly delayed tumor growth at the injected site compared with the succinate and empty VLP controls (figure 3A), which was more clearly observed in the spaghetti plots shown in figure 3C–F. Of note, empty VLPs induced significant suppression of tumor growth compared with succinate in the treated tumors (figure 3A), suggesting that the VLP itself may trigger an immune response. CMP-001 was more efficient at controlling distant (uninjected) tumor growth (6 out of 10 tumors regressed, figure 3B,F) and at improving survival (figure 3G) than G10 (2 out of 10 tumors regressed, figure 3B,E). Mice primed with CMP-001 developed a robust anti-Qβ response after 2 weeks in contrast to mice primed with empty VLP, G10 and succinate buffer (figure 3H). These results suggest that encapsulation of G10 in the VLP (CMP-001) is important for inducing both a favorable local and a systemic antitumor immune response thus leading to superior survival outcomes.

CMP-001 showed superior antitumor effect in vivo compared with G10. mEERL tumor-bearing C57BL/6 mice (n=10/treatment group) were treated intratumorally with succinate as a control, G10 CpG ODN, empty virus-like particle (VLP) (100 µg/mouse) and CMP-001 (100 µg/mouse) into the left tumor on Days 14, 17 and 20 after mEERL tumor inoculation. All mice were primed 2 weeks before tumor inoculation with their respective assigned treatments. Tumor volume for the left (injected) (A) and right (distant (uninjected)) (B) tumors in the treated mice was measured three to four times per week. Tumor growth curves shown were terminated when a mouse in any treatment group reached euthanasia criteria. Spaghetti plots of all mice in each of the treatment groups are shown in (C)–(F). Numbers indicated in the spaghetti plots are the number of distant tumors that regressed. Overall survival was analyzed by constructing Kaplan-Meier plots (G) for the treated mice. Mouse serum from a subset of mice (n=4–5 mice/treatment group) was collected 2 weeks after priming for analysis of immunoglobulin (Ig) anti-Qβ concentration (H). Error bars represent SD from the mean. *p<0.05. NS, non-significant.
CMP-001 significantly enhances anti-PD1 therapy in vivo
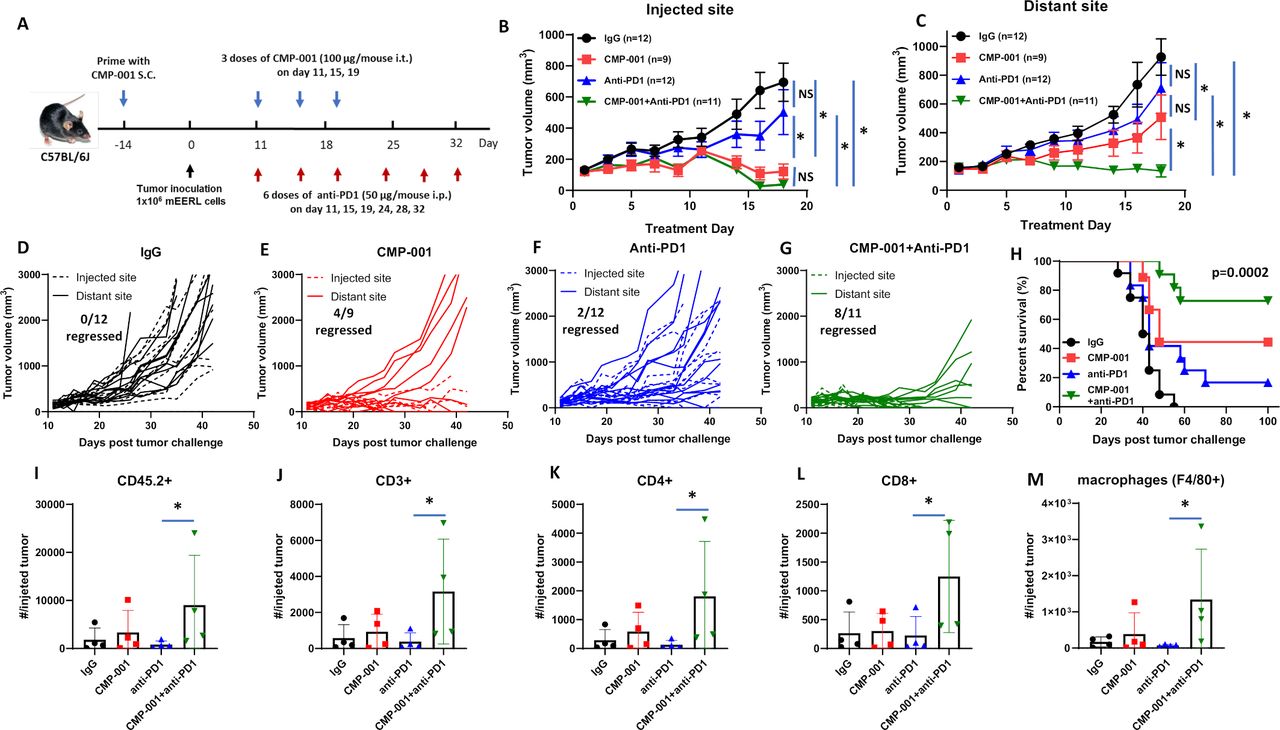
We next investigated if CMP-001 could enhance tumor response to anti-PD-1 therapy. mEERL-bearing mice were administered CMP-001 on Days 11, 15 and 19 after tumor cell inoculation to the left tumor with or without anti-PD-1 intraperitoneally two times per week for a 3-week treatment period, as illustrated in figure 4A. Isotype control was administered intraperitoneally as a control. All mice receiving CMP-001 were primed with CMP-001 2 weeks before tumor inoculation. Results showed the CMP-001 alone (figure 4B,E) and in combination with anti-PD-1 (figure 4B,G) significantly suppressed tumor growth at the injected tumor compared with control (figure 4B,D). CMP-001 alone (figure 4C,E) and CMP-001+anti-PD-1 (figure 4C,G) also significantly suppressed distant tumor growth compared with control (figure 4C,D). However, only CMP-001+anti-PD-1 significantly suppressed distant tumors (8/10 tumors regressed) compared with control (0/12 tumors regressed, figure 4C,D), CMP-001 (4/9 tumors regressed, Figure 4C,E) and anti-PD-1 (2/12 tumors regressed, figure 4C,F). CMP-001+anti-PD-1 also significantly enhanced survival (median survival=not reached) compared with CMP-001 (median survival=48 days), anti-PD-1 (median survival=43 days) or control group (median survival=41 days) (figure 4H). These results suggest that CMP-001+anti-PD-1 therapy can target both local and systemic disease and thus improve survival outcomes.

CMP-001 enhances anti-programmed cell death protein-1 (anti-PD1) therapy. (A) mEERL tumor-bearing C57Bl/6 mice (n=9–12/treatment group) were treated with immunoglobulin G (IgG) (+succinate) as a control, CMP-001 (+IgG), anti-PD1 (+succinate) and CMP-001+anti-PD1. CMP-001 and succinate were administered intratumorally into the left tumor on Days 11, 15, and 19, and anti-PD1 was administered intraperitoneally on Days 11, 15, 19, 24, 28, and 32 after mEERL tumor inoculation. All mice receiving CMP-001 were primed 2 weeks before tumor innoculation. Tumor volume for the left (injected) (B) and right (distant (uninjected)) (C) tumors in the treated mice was measured three to four times per week. Tumor growth curves shown were terminated when a mouse in any treatment group reached euthanasia criteria. Spaghetti plots of all mice in each of the treatment groups are shown in (D)–(G) Numbers indicated in the spaghetti plots are the number of distant tumors that regressed. Overall survival was analyzed by constructing Kaplan-Meier plots (H) for the treated mice. A subset of mice (n=3–4 mice/group) from each treatment were euthanized 2 days after their last treatment, and tumors at the injected tumor site were harvested and analyzed by flow cytometry for select immune subsets such as CD45.2+ cells (I), CD3+ (J), CD4+ (K), and CD8 + (L) T cells and macrophages (M). Error bars represent SD from the mean. *p<0.05. NS, non-significant.
CMP-001+ anti-PD-1 increases immune cell infiltration to the draining LN
To assess changes in immune cell infiltration and activation, tumors and lingual draining LNs at the injected site were harvested from mice 2 days after treatment completion from figure 4A–C and analyzed by flow cytometry for differences in immune cell subsets among the treatment groups. In tumors, significant differences were observed between anti-PD-1 and CMP-001+anti-PD-1-treated tumors for CD45.2+ (figure 4I), CD3+ (figure 4J), CD4+ (figure 4K), CD8+ (figure 4L) and macrophages (figure 4M). In draining LNs, there was a general increase in DCs (figure 5A), monocytes (figure 5B), T cells (figure 5C–G) and HPV+ T cells (figure 5H) due to CMP-001+ anti-PD-1 treatment compared with control. However, significant differences were only observed with CD3+ (figure 5C), CD4+ (figure 5D) and CD8+ T cells (figure 5F) compared with control. CMP-001+anti-PD-1 also significantly increase IFNγ+ CD4+ (figure 5E) and IFNγ+ CD8+ T cells (figure 5G) compared with anti-PD-1 alone. Circulating levels of cytokines/chemokines were also analyzed from a subset of mice 24 hours after treatment. Significant increases in TNFα (figure 5I), IL-6 (figure 5J), IL-5 (figure 5K) and IL-12 (p70) (figure 5L) levels were observed in CMP-001+anti-PD-1-treated mice compared with control-treated mice. Circulating TNFα and IL-6 levels were also significantly increased in CMP-001+anti-PD-1-treated mice compared with anti-PD1 alone (figure 5I,J). These results suggest that CMP-001 may enhance tumor response to anti-PD-1 therapy by increasing the recruitment of activated T cells to the draining LN.

CMP-001 combined with anti-programmed cell death protein-1 (anti-PD1) increases immune cell infiltration to the draining lymph node (LN) . A subset of mice (n=3–4 mice/group) from each treatment group from the experiment shown in figure 4A–C were euthanized 2 days after their last drug treatment and lingual LNs at the injected tumor site were harvested and analyzed by flow cytometry for select immune subsets such as dendritic cells (DCs) (A), monocytes (B), CD3+ (C), CD4+ (D), IFNγ+ CD4+ (E), CD8+ (F), IFNγ+ CD8+ (G) and HPV+ CD8+ (H) T cells. Serum samples were collected for cytokine analysis using a murine cytokine multiplex assay. Shown are concentration (µg/mL) levels for TNFα (I), IL-6 (J), IL-5 (K) and IL-12(p70) (L). Error bars represent SD from the mean. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. HPV+, human papilloma virus-positive; IFNγ, interferon gamma; IgG, immunoglobulin G; IL, interleukin.
The antitumor effects of CMP-001+ anti-PD1 are CD8+ T cell dependent
Given that CMP-001+ anti-Qβ was successful at increasing the activation of CD4+/CD8+ T cells and NK cells in vitro (figure 1), we investigated the impact of depleting each of these immune cell subsets on tumor response to CMP-001+anti-PD-1 in vivo. Depletion of CD8+ T cells but not CD4+ T cells significantly and completely protected injected (figure 6A) and distant (figure 6B) tumors from the antitumor effects of CMP-001+anti-PD-1 and significantly reduced survival (figure 6C) compared with CMP-001+anti-PD-1 without immune cell depletion. NK cell depletion also significantly protected injected (figure 6A) and distant (figure 6B) tumors from CMP-001+anti-PD-1 but there was no difference in overall survival of mice between anti-NK+CMP-001+anti-PD-1 and CMP-001+anti-PD-1-treated mice (figure 6C). Validation of CD4+/CD8+ T cells and NK cells is shown in figure 6D–F. These results suggest that CD8+ T cells play a central role in CMP-001+anti-PD-1-induced antitumor responses and survival outcomes.
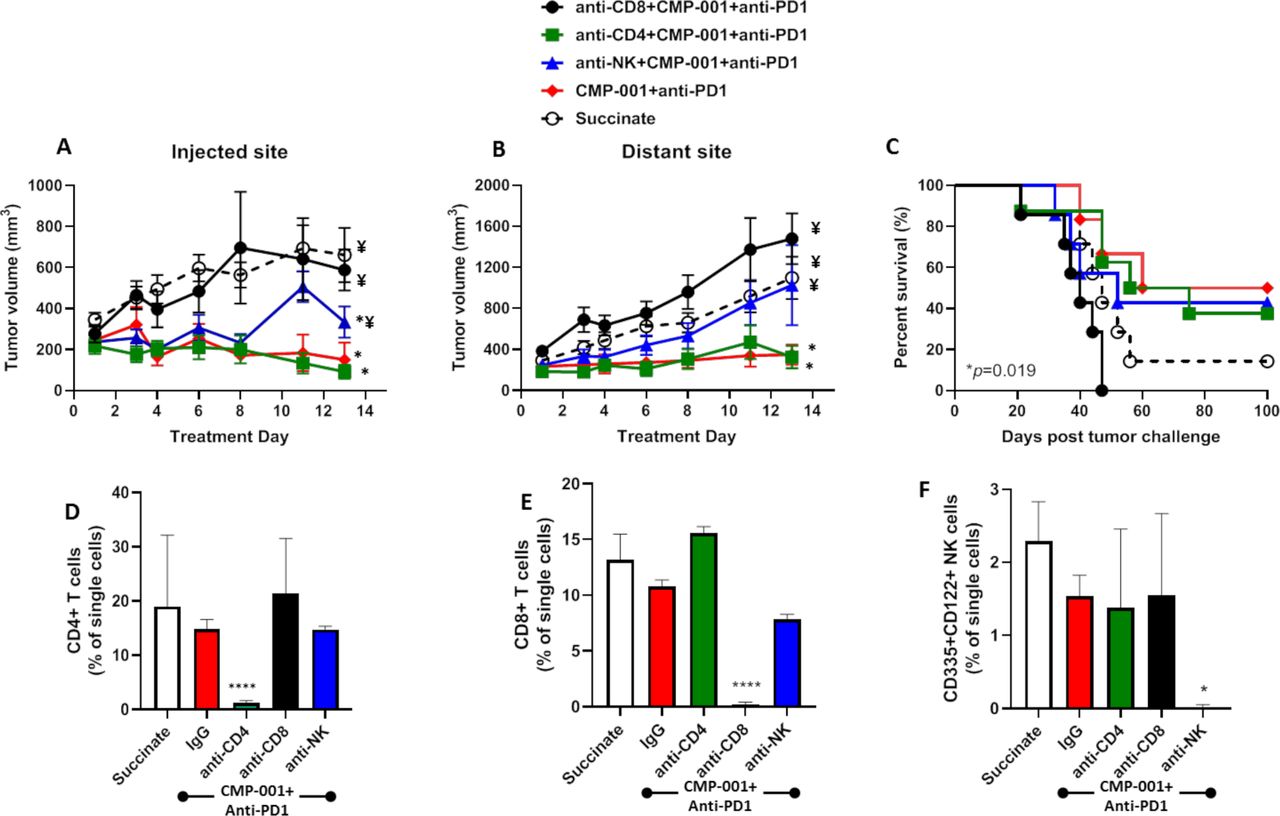
The antitumor effects of CMP-001+α-PD1 depends on CD8+ T cells. mEERL tumor-bearing C57Bl/6 mice (n=10–12/treatment group) were treated with CMP-001+anti-programmed cell death protein-1 (anti-PD1) as described in figure 4 with or without anti-CD4, anti-CD8 or anti–asialo-GM1 (anti-NK) antibodies 1 and 3 days prior to tumor inoculation, and every 3–4 days after tumor inoculation. Succinate buffer was used as non-treatment control. All mice receiving CMP-001 were primed 2 weeks before tumor inoculation. Tumor volume for the left (injected) (A) and right (distant (uninjected)) (B) tumors in the treated mice was measured three to four times per week. Tumor growth curves shown were terminated when a mouse in any treatment group reached euthanasia criteria. Overall survival was analyzed by constructing Kaplan-Meier plots (C) for the treated mice. *p<0.05 versus succinate, ¥p<0.05 versus CMP-001+ anti-PD1. (D–F) Splenic peripheral blood mononuclear cells (PBMCs) were isolated from a subset of mice (n=3) 1 day after the last treatment and were analyzed by flow cytometry for validation of CD4+ T cell (D), CD8+ T cell (E), and natural (NK) cell (F) depletion. *p<0.05; ****p<0.0001. Error bars represent SD from the mean. IgG, immunoglobulin G; NK, natural killer.
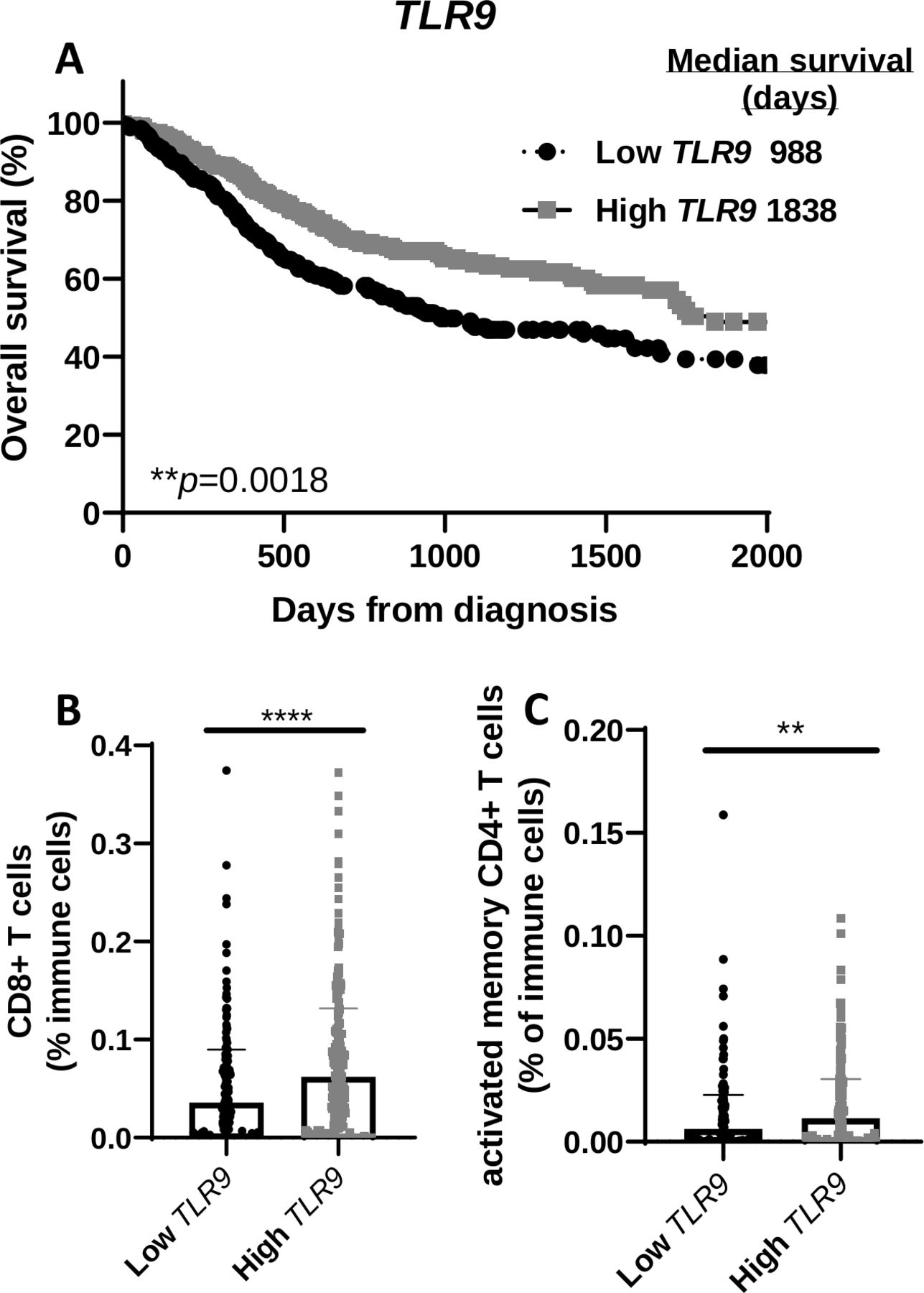
High expression of TLR9 correlates with higher overall survival in patients with HNSCC
To determine any clinical relevance of TLR9 gene expression, patients with HNSCC from the TCGA database were divided into two groups based on TLR9 expression levels and labeled as ‘high’ (n=284) and ‘low’ (n=236). Survival curves showed that HNSCC patients with higher levels of TLR9 expression (median survival=1838 days) were associated with a more favorable survival compared with patients with lower TLR9 expression levels (median survival=988) (figure 7A). There was no significant difference between the two TLR9 expression groups with regard to available patient characteristics in the TCGA database (eg, gender, age, clinical stages, primary/follow-up treatment success, online supplemental table 1). Further analysis of levels of select immune cells in tumors from patients with HNSCC in the two TLR9 expression groups showed that tumors in the high TLR9 expression group exhibited significantly higher CD8+ T cells (figure 7B) and activated memory CD4+ T cells (figure 7C) compared with the low TLR9 expression group. These results suggest that the favorable survival observed in patients with higher TLR9 expression may be due to the presence of tumor-specific T cells.
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TLR9 gene expression positively correlates with overall survival of patients with head and neck squamous cell carcinoma (HNSCC) and T cell infiltration. (A) Kaplan-Meier survival curves comparing overall survival of patients with HNSCC (n=520) created from The Cancer Genome Atlas (TCGA) database according to high (n=284) and low (n=236) TLR9 tumor gene expression. (B, C) Dot plots illustrate the percentage of CD8+ (B) and activated memory CD4+ (C) tumor-infiltrating T cells in HNSCC patients with high and low TLR9 expression from (A). Error bars represent SD from the mean. **p<0.01; ****p<0.0001.
Discussion
Our work highlights CMP-001 as a promising adjuvant to anti-PD1 therapy for HNSCC to target both local and distant tumors, which supports prior work with TLR9 agonists in melanoma,17 18 27 lymphoma15 and other models.28 29 The majority of patients with HNSCC (50%–60%) will be diagnosed with local invasion and regional LN metastases, and 15%–25% will be found with distant metastases after standard therapy (radiotherapy±chemotherapy).30 Therefore, this drug combination may improve locoregional control and target undetected distant tumors in patients with HNSCC which would be a significant advancement for HNSCC therapy.
As expected, CMP-001 directly activated pDCs (figure 1A,B) which likely led to T cell activation when in PBMCs/HNSCC cell line co-cultures (figure 1E,F). This is supported by the observation that isolated T cells could not be directly activated by CMP-001 (data not shown). CMP-001 also activated NK cells in PBMCs/HNSCC cell line coculture systems (figure 1G) which could be explained by DC-NK cell crosstalk.31 NK cells could be also directly activated by CMP-001 since isolated NK cells were able to be activated (online supplemental figure 1), although to a lesser extent than when DCs were present. There is a controversy about whether NK cells express TLR9 or not. It is possible that the Fc portion of anti-Qβ could be engaging the Fc receptor on NK cells which would induce NK cell activation independent of TLR9 activation. However, TLR9 expression in NK cells has been confirmed in a number of studies,32–34 NK cells have been found to respond to CpG ODNs, and NK cells from TLR9-deficient mice are unable to respond to CpG ODNs.32 The direct and indirect effects of CMP-001 on NK cell activation suggest that CMP-001 may also be a promising adjuvant to agents that trigger NK cell activity and ADCC such as cetuximab and trastuzumab. In fact, the TLR9 agonist immunomodulatory oligonucleotide was reported to enhance cetuximab activity in K-ras mutant colorectal and pancreatic cancers16 and enhance trastuzumab in breast cancer.35
It is clear from our work that anti-Qβ boosts the antitumor immune response to CMP-001 (figure 2). Anti-Qβ is necessary for opsonization of the VLP, which allows for uptake by pDCs in vitro. Specifically, anti-Qβ is believed to form an immune complex with CMP-001, and the Fc portion of anti-Qβ binds to the Fc receptor (FcR) on pDCs, facilitating phagocytosis and subsequent pDC activation. This is supported by prior work showing that blocking the FcR, using an FcR-blocking Ab (anti-CD32), significantly reduced production of IFNα by PBMCs stimulated with CMP-001+anti-Qβ.15 Given that it took about 2 weeks to observe robust anti-Qβ development after CMP-001 administration in vivo, and the rapid growth of mEERL tumors in our in vivo model, it was necessary to include a 2-week ‘prime’ step in the experimental design, similar to what was done in a previous work with lymphoma.15 It is recognized that a ‘priming’ step would not be relevant for patients with cancer since they would have pre-existing tumors. However, since human tumors grow at a much slower rate than tumors implanted in mice, there would be an appropriate period of time to develop an anti-Qβ response after the initial CMP-001 administration. The ‘prime’ step used in our preclinical experiments is thus important for achieving the desired circulating anti-Qβ levels in order to study the long-term effects of CMP-001 before tumors reach euthanasia criteria (≥15 mm).
The incidence of HPV‐positive HNSCC tumors is increasing worldwide, and is characterized by a generally less aggressive phenotype and is associated with a more favorable prognosis compared with HPV- tumors.36 37 In the in vitro co-culture assays, we observed significantly more immune cell stimulation and cytokine secretion with the HPV+ cell line compared with the HPV- cell line (figure 1E–I). HPV-driven cancers present viral antigens that elicit HPV-oncoprotein-specific antibodies as well as T cell responses, as shown in figure 5H, suggesting that HPV+ tumors may be more immunogenic than HPV- tumors38 and are more responsive to immunotherapy including TLR9 agonists. This difference in immunogenicity may explain the differences in immune cell activation observed between HPV+and HPV- coculture systems (figure 1E–I). It is unclear if these differences can be solely attributed to HPV status since the two HNSCC cell lines are different; however, future work will determine if HPV+ tumors are more responsive to CMP-001+ anti-PD1 than HPV- tumors.
In conclusion, the work presented here provides a rationale for the use of CMP-001 to activate pDCs in HNSCCs, which would trigger type I IFN production, stimulate immune cell trafficking into draining lymph nodes and tumors, and possibly increase the immunogenicity of HNSCC tumors. This would allow for enhanced systemic antitumor immune responses and increased durable responses when combined with immune checkpoint therapy.
Acknowledgments
The authors thank Checkmate Pharmaceuticals, Cambridge, Massachusetts (USA) for providing the CMP-001 and acknowledge Drs. Douglas E. Laux, Jon Houtman, Gabriele Ludewig and Thomas Waldschmidt for their helpful discussions regarding the work shown in this manuscript, and the DeGowin Blood Center at the University of Iowa Hospitals and Clinics. The data presented herein were obtained at the Flow Cytometry Facility, which is a Carver College of Medicine/Holden Comprehensive Cancer Center core research facility at the University of Iowa. The facility is funded through user fees and the generous financial support of the Carver College of Medicine, Holden Comprehensive Cancer Center and Iowa City Veteran's Administration Medical Center.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Conception and design: YC, AS, CL-M, GJW. Development of methodology: YC, CL-M. Acquisition of data: YC, WW, ZW. Analysis and interpretation of data: YC, CL-M, AS, ZW, CC, AS, GJW. Writing, review, and/or revision of the manuscript: YC, CL-M, CC, AKS, GJW, AS. Study supervision: AS.
Funding Research supported by the Mezhir Award Program through the Holden Comprehensive Cancer Center and National Institutes of Health (NIH) grant R01DE024550 (ALS).
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Data are available upon reasonable request. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.