Article Text

Abstract
Background Adoptive cell therapy with chimeric antigen receptor T cells (CAR-T) has become a standard treatment for patients with certain aggressive B cell malignancies and holds promise to improve the care of patients suffering from numerous other cancers in the future. However, the high manufacturing cost of CAR-T cell therapies poses a major barrier to their broader clinical application. Among the key cost drivers of CAR-T production are single-use reagents for T cell activation and clinical-grade viral vector. The presence of variable amounts of contaminating monocytes in the starting material poses an additional challenge to CAR-T manufacturing, since they can impede T cell stimulation and transduction, resulting in manufacturing failure.
Methods We created K562-based artificial antigen-presenting cells (aAPC) with genetically encoded T cell stimulation and costimulation that represent an inexhaustible source for T cell activation. We additionally disrupted endogenous expression of the low-density lipoprotein receptor (LDLR) on these aAPC (aAPC-ΔLDLR) using CRISPR-Cas9 gene editing nucleases to prevent inadvertent lentiviral transduction and avoid the sink effect on viral vector during transduction. Using various T cell sources, we produced CD19-directed CAR-T cells via aAPC-ΔLDLR-based activation and tested their in vitro and in vivo antitumor potency against B cell malignancies.
Results We found that lack of LDLR expression on our aAPC-ΔLDLR conferred resistance to lentiviral transduction during CAR-T production. Using aAPC-ΔLDLR, we achieved efficient expansion of CAR-T cells even from unpurified starting material like peripheral blood mononuclear cells or unmanipulated leukapheresis product, containing substantial proportions of monocytes. CD19-directed CAR-T cells that we produced via aAPC-ΔLDLR-based expansion demonstrated potent antitumor responses in preclinical models of acute lymphoblastic leukemia and B-cell lymphoma.
Conclusions Our aAPC-ΔLDLR represent an attractive approach for manufacturing of lentivirally transduced T cells that may be simpler and more cost efficient than currently available methods.
- receptors, chimeric antigen
- immunotherapy, adoptive
- antigen presentation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Chimeric antigen receptor T-cell (CAR-T) therapy has revolutionized the treatment of hematological malignancies. CAR-T cells are a form of adoptive immunotherapy that reprograms a patient’s T-cells to target malignant cells based on their expression of tumor-specific or tumor-associated surface antigens. CD19-directed CAR-T therapy has quickly advanced and now is an Food and Drug Administration (FDA)-approved treatment for children and young adults with relapsed/refractory B-cell acute lymphoblastic leukemia (ALL) and adults with relapsed/refractory large B-cell lymphoma.1 Promising results have also been obtained from early-phase clinical trials using CD22-directed CAR-T against B-cell malignancies2 and B cell maturation antigen (BCMA)-targeting CAR-T for the treatment of multiple myeloma.3 Although CAR-T therapy for solid cancers has not yet been able to match the impressive success achieved by their hematological counterparts, encouraging results have been reported for some solid tumors.4 With more than 300 clinical CAR-T trials underway worldwide for numerous hematological and solid tumors,5 CAR-T therapy is promising to a larger number of patients in the future.
One of the major barriers to broader implementation of the engineered T cells as a therapeutic platform is the high cost of bespoke manufacturing, including the need for Good Manufacturing Practices (GMP)-grade, single-use reagents that are in limited supply and each have associated high costs. Furthermore, even in the hands of experienced manufacturers, CAR-T production at a commercial scale fails in up to 10% of cases6 due to the varying composition of the starting material used to manufacture CAR-T cells.7 Most methods of generating CAR-T use soluble or bead-bound antibodies against CD3 and CD28 along with IL-2 supplementation to activate the T cells, each of which must be generated and secured according to GMP to meet regulatory requirements for clinical use. Activated T cells are then transduced with retroviral or lentiviral vectors that encode the CAR or desired transgene.8
Renewable stocks of artificial antigen-presenting cells (aAPC) from a working cell bank can also be used to activate T cells prior to transduction. K562-based aAPC can be used off the shelf and represent an inexhaustible, cost-efficient, single reagent for T cell expansion. K562, a human myelogenous leukemia cell line, are an attractive scaffold for the construction of cell-based aAPC because they lack expression of human leukocyte antigen (HLA) class I and HLA class II molecules, as well as costimulatory or coinhibitory molecules, making them unlikely to induce unwanted allospecific T cells.9 The safety of using irradiated K562 cells in human subjects has also been previously demonstrated.10 11 However, one drawback of using K562-based aAPC is their susceptibility to transduction by lentiviral vectors due to their constitutive expression of the low-density lipoprotein receptor (LDLR) that serves as the entry receptor for Vesicular stomatitis virus-G (VSV-G) pseudotyped vectors.12 13 Inadvertent transduction of the aAPC could reduce transduction of T cells, or could confer undesirable biology on the aAPC.
In this study, we developed a self-contained cell-based aAPC reagent that does not require use of any soluble antibodies to manufacture CAR-T cells. We transduced K562 cells with T cell stimulatory receptors and we achieved genetic resistance to lentiviral transduction via knockout of LDLR (aAPC-ΔLDLR). We demonstrate that our aAPC-ΔLDLR efficiently produce CD19-targeting CAR-T that display potent antitumor efficacy in preclinical models of ALL and B-cell lymphoma. In addition, we found that when the starting material contained monocytes, as is typical for peripheral blood mononuclear cells (PBMC) or unmanipulated whole leukopaks, our aAPC-ΔLDLR-based approach was less prone to variability in CAR-T expansion compared with a standard bead-based approach.
Material & methods
Cell lines and culture condition
K562, Nalm6 and Jeko-1 cell lines were purchased from the American Type Culture Collection. Nalm6 and Jeko-1 cell lines were transduced to stably express click beetle green luciferase (CBG-luc). Cells expressing the desired phenotype were bulk sorted on a FACSAria (BD Biosciences) to obtain a≥ 99% pure population. All cell lines were cultured in RPMI media (10% FBS, 1% penicillin/streptomycin).
Optimizing LDLR knockout and analysis by T7E1 assay
To identify highly active combinations of CRISPR-Cas nucleases and guide RNAs capable of knocking out the LDLR in K562 cells, a series of transfections were performed. Plasmids encoding Cas9 and sgRNA (750 ng and 250 ng, respectively) or Cas12a and crRNA (580 ng and 250 ng, respectively) were electroporated into approximately 2×105 K562 cells via the FF-120 program with the SF Cell Line Nucleofector Kit using a 4D-Nucleofector (Lonza). Genomic DNA was extracted approximately 72 hours post nucleofection using the Agencourt DNAdvance Nucleic Acid Isolation Kit (Beckman Coulter). Analysis of on-target editing was performed by T7 Endonuclease I (T7EI) assay, similar to as previously described.14 Briefly, the locus surrounding the on-target site was PCR amplified from ~100 ng of genomic DNA as template, and ~200 ng of purified PCR products were denatured, annealed, digested with 10 U T7EI (New England Biolabs) at 37°C for 25 min, and cleaned up using AMPureXP beads (Beckman Coulter). The digests were analyzed using a QIAxcel (Qiagen) to estimate target site modification, with total editing calculated as 100 × (1 – (1 – fraction cleaved)1/2).
Construction of K562-based aAPC
Two chimeric stimulatory receptors (CSRs), α-CD3-GFP and α-CD28-mCherry, were designed based on an α-CD3 single-chain variable fragment (scFv) derived from the OKT3 clone and an α-CD28 scFv derived from the 9.3 clone, respectively. aAPC were then generated from wildtype K562 cells by lentiviral introduction of the two CSRs, and subsequent bulk sorting for GFP+ mCherry+ cells. Next, CRISPR-SpCas9 was used to generate the aAPC-ΔLDLR cell line with disruption of the LDLR gene. Single-cell clones were sorted based on GFP, mCherry, and LDLR expression. aAPC cultures derived from the same single-cell clone were used for all subsequent assays.
aAPC irradiation and transductions
aAPC were irradiated at 100 gray using a Cesium-137 irradiator and cryopreserved immediately thereafter. For experiments testing aAPC’s transduction susceptibility, aAPC were thawed and cultured for 24 hours prior to adding blue fluorescent protein (BFP) encoding lentivirus. In experiments using receptor-associated protein (RAP; Enzo Life Sciences), cultures were incubated with RAP (50 nM, 15 min, 37°C) prior to lentiviral infection. At timepoints specified per experiment, transduction susceptibility was measure by flow cytometry based on BFP expression level.
T cell expansion and CAR-T production
Primary human blood cells were obtained and cryopreserved under an IRB-approved protocol using anonymous healthy donor leukopaks purchased from the MGH blood bank. As starting material for CAR-T manufacturing, either purified T cells (Stem Cell Technologies, Cat#15061), isolated PBMC, or whole unmanipulated leukopak was used as specified in the legends. In experiments using whole unmanipulated leukopak as starting material, the T cell number was estimated based on the absolute lymphocyte count measured on an ABX Micros 60 instrument (Horiba). For expansion and CAR-T production, T cells or PBMC were thawed and activated using either α-CD3/α-CD28 Dynabeads (Life Technologies) at a 1:3 T cell:beads ratio, or freshly thawed irradiated aAPC-WT or aAPC-ΔLDLR at a 2:1 T cell:aAPC ratio. The T cell:aAPC ratio of 2:1 was chosen based on prior experience with K562-based aAPCs.15 16 The cells were cultured in RPMI medium (10% FBS, 1% penicillin/streptomycin) supplemented with 20 IU/mL of rhIL-2. Every other day, fresh media was added to keep the cells at a concentration of 0.5–2×106/mL. For CAR-T production, lentiviral vector was added to the culture 24 hours post activation (multiplicity of infection 1–5). In parallel, donor-matched T cells that had been activated but untransduced (UTD) were expanded to serve as a negative control in subsequent experiments. At days 12–14 of culture, CAR expression was determined and normalized across CAR-T products by adding UTDs prior to cryopreservation.
To calculate population doublings, the following equation was used: log2(product of increase factors day0 to dayn), where increase factor = (total # cells dayn/total # of cells dayn–1).
Flow cytometry
Antibodies used: α-human CD3 APC (Clone OKT3, BioLegend), α-human CD3 BUV395 (Clone UCHT1, BD Biosciences), α-human CD3 BV421 (Clone OKT3, BioLegend), α-human CD3 FITC (Clone UCHT1, BioLegend), α-human CD4 BV510 (Clone SK3, BD Biosciences), α- human CD4 BV786 (Clone SK3, BD Biosciences), α-human CD8 APC-H7 (Clone SK1, BD Biosciences), α-human CD8 BUV395 (Clone RPA-T8, BD Biosciences), α-human CD69 APC (Clone FN50, BioLegend), α-human CD107a Alexa Fluor 700 (Clone H4A3, BD Biosciences), α-human LDLR APC (Sino Biological), α- mouse/human CD11b-APC (Clone M1/70, BioLegend), α-mouse Ly-6G/Ly-6C(Gr-1)-APC (Clone RB6-8C5, BioLegend), α-mouse NK-1.1-APC (Clone PK136, BioLegend), and α-mouse TER-119/erythroid cells-APC (Clone TER-119, BioLegend).
Cells were stained for 20 min at 4°C protected from light, washed twice in phosphate buffered saline (PBS) with 2% FBS, acquired on a Fortessa X-20 (BD Biosciences), and analyzed in FlowJo software. Assays to measure activation of T cells in vitro, as well as quantification of T cell persistence in treated mice, were performed as has been previously described.17
Cellular cytotoxicity
Cocultures of normalized CAR-T with CBG-luc-expressing Nalm6 and Jeko-1 cells were set up at the indicated effector:target (E:T) ratios for 8 hours. Luciferase activity was measured using a luciferase assay system (Promega, Cat# E1501) according to manufacturer’s protocol and a Synergy Neo2 luminescence microplate reader (Biotek). Percent specific lysis was calculated by the following formula: percent specific lysis = ((target cell only relative light units (RLU) – well of interest RLU)/target cell only RLU)×100.
In vivo studies
All animal procedures were performed in accordance with Federal and Institutional Animal Care and Use Committee requirements under a protocol approved at Massachusetts General Hospital. NOD-SCID-γ chain−/− (NSG; Jackson Laboratories) were engrafted with 1×106 Nalm6 cells, transduced to express CBG-luc, via intravenous injection. Seven days after tumor injection, once engraftment of tumor was confirmed by bioluminescence imaging (BLI), 2×106 cryopreserved CAR-19 T cells or T cells, that had been activated but not transduced (UTD), were injected intravenously. Tumor burden was monitored using an Ami spectral imaging apparatus and analyzed with IDL software V.4.3.1. Animals were euthanized when they met prespecified endpoints defined by the IACUC or at the end of an experiment.
Statistics
All statistical analyses were performed using GraphPad Prism V.8.4.1. Unless otherwise indicated, a two-tailed Student’s t-test or two-way analysis of variance test was used for normal data at equal variance. P<0.05 was considered significant and is marked as * in the legends. T cells from at least three different healthy donors were used for all in vitro and in vivo experiments. For specific numbers of technical replicates and biological replicates, refer to the legends.
Results
K562-based aAPC lacking expression of the LDLR are resistant to VSV-LV transduction
We constructed two CSR employing an α-CD3 scFv coupled with a GFP reporter and an α-CD28 scFv coupled with a mCherry reporter (figure 1A,B). These CSRs were transfected into K562 cells. Stable expression of both the α-CD3 CSR and the α-CD28 CSR in the newly generated aAPC was confirmed and single-cell clones were generated by fluorescent-activated cell sorting (FACS; online supplementary figure 1). Next, to identify the optimal conditions for disruption of the LDLR gene in K562 cells, we tested the activities of SpCas9, SpCas9-HF1,18 AsCas12a, and LbCas12a19 when targeted to several sites in exons 1 and 3 (online supplementary figure 2). Use of SpCas9 to target site 2 in exon 1 of the LDLR gene showed the best knockout activity in K562 cells and was chosen for subsequent generation of K562LDLR KO cells. Stable knockout of the LDLR was confirmed and single-cell clones were generated by FACS (figure 1C).
Supplemental material
Supplemental material
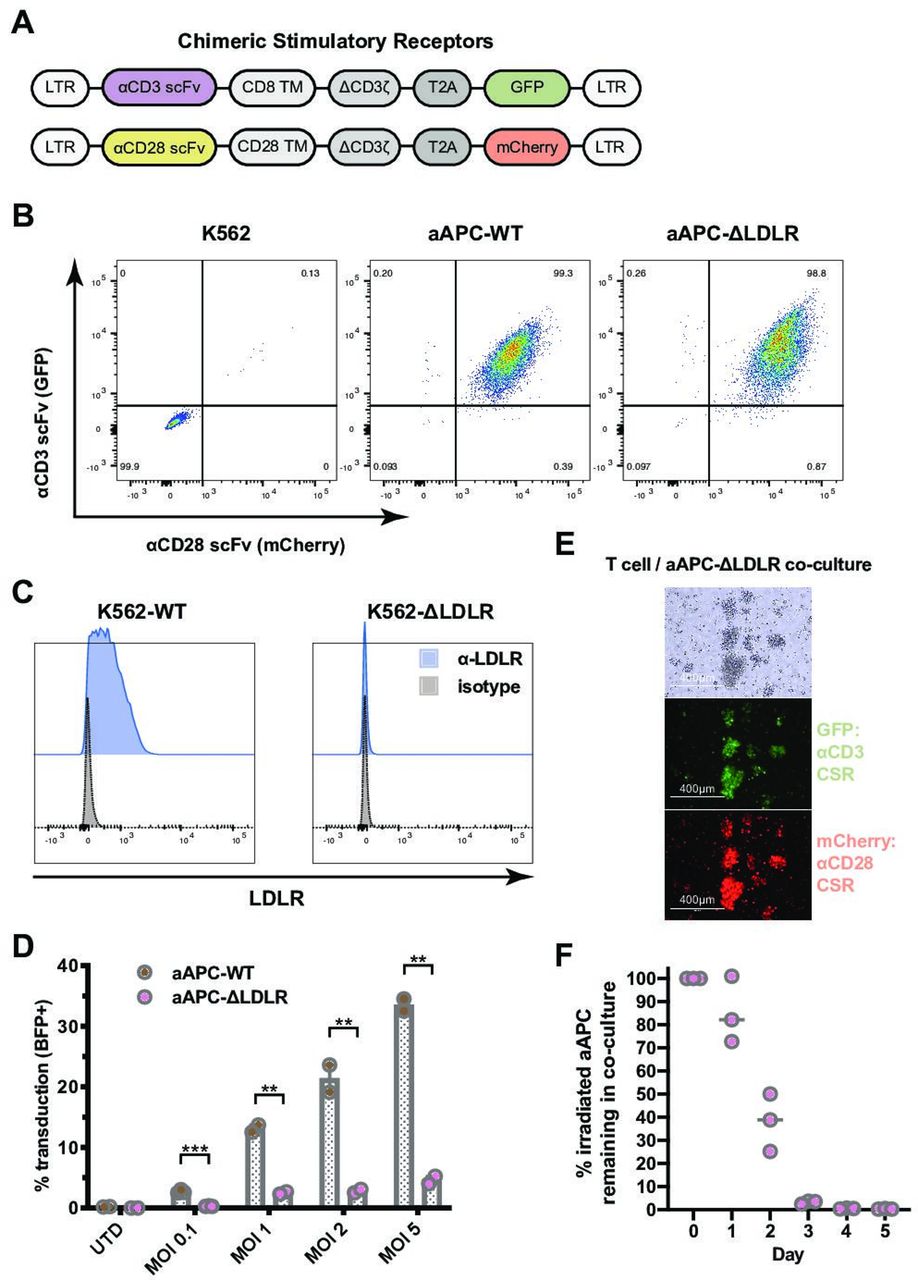
K562-based artificial antigen-presenting cells (aAPC) that lack expression of the low-density lipoprotein receptor (LDLR) are resistant to VSV-G pseudotyped lentiviral vector (VSV-LV) transduction. (A) Two chimeric stimulatory receptors (CSR) were constructed: one employing an α-CD3 single-chain variable fragment (scFv) and a GFP reporter (top) and one with an α-CD28 scFv and an mCherry reporter (bottom). LTR, long terminal repeat; TM, transmembrane domain; T2A, 2A self-cleaving peptide. (B) Stable expression of both the α-CD3 CSR and the α-CD28 CSR on the newly generated aAPC, aAPC-WT and aAPC-ΔLDLR, was confirmed by flow cytometry. (C) The LDLR was knocked out from the parental K562 cell line using CRISPR/Cas9 gene editing. Lack of LDLR surface expression on the newly generated K562-ΔLDLR was confirmed by flow cytometry. (D) Susceptibility of irradiated aAPC-WT and aAPC-ΔLDLR to VSV-LV transduction was assessed using a blue fluorescent protein (BFP)-encoding lentiviral vector at various multiplicities of infection. The percentage of BFP+ aAPC (GFP+mCherry+) was measured after 48 hours by flow cytometry. Bars show mean ± SEM of experiment performed in triplicate. Unpaired, two-tailed t-test, ***p ≤ 0.001, **p ≤ 0.01. (E) and (F) Duration of stimulatory signal provided by aAPC-ΔLDLR in coculture with T cells. (E) Bright-field and fluorescence microscopy images of T cells and aAPC-ΔLDLR that have been cocultured for 18 hours. (F) Disappearance of irradiated aAPC-ΔLDLR in coculture with T cells over time. Horizontal line indicates mean of experiment performed in triplicate.
We proceeded to test the hypothesis that knockout of LDLR reduces aAPC’s susceptibility to transduction via VSV-G pseudotyped lentiviral vector (VSV-LV). Susceptibility of irradiated aAPC-WT and aAPC-ΔLDLR to VSV-G infection was assessed using a BFP-encoding lentiviral vector at various multiplicities of infection (MOIs). The percentage of BFP+ aAPC (GFP+ mCherry+) was measured after 48 hours by flow cytometry. Across all MOIs tested, we found that knockout of the LDL receptor significantly reduced aAPC susceptibility to VSV-LV transduction by a factor of up to 7 (p=0.002; figure 1D). Importantly, using MOIs within the clinically relevant range of 0.1–1, the residual transduction of aAPC-ΔLDLR at 0.3% and 2%, respectively, was negligible. In contrast, when using high MOIs we observed low-level residual transduction of aAPC-ΔLDLR and therefore interrogated whether other LDLR family members can serve as alternative entry receptors for VSV-LV, as has been reported for other cell types.12 20 To investigate alternative entry mechanisms for VSV-LV in the context of our K562-based aAPC, we made use of RAP, a molecular chaperon that is required for the maturation of LDLR family members. Physiologically, RAP acts by binding LDLR family members and preventing their premature interaction with ligands during the secretory process.21 Since RAP blocks ligand binding to all members of the LDLR family except for LDLR itself, we used it to examine potential alternative mechanisms of VSV-LV entering K562-based aAPC.12 22 Preincubation of aAPC-WT and aAPC-ΔLDLR with RAP did not alter their susceptibility to transduction with a BFP-VSV-LV vector across various MOIs (online supplementary figure 3), indicating that K562 cells do not express alternative entry receptors for VSV-LV at a biologically relevant level. Therefore, knockout of the LDLR on K562 cells is sufficient to make them resistant to lentiviral transduction in a clinically relevant manufacturing setting.
Supplemental material
One important consideration when using aAPC is the duration of stimulation that the T cell will receive. Natural T cell activation is triggered by short-term antigen presentation. Sustained TCR signaling can induce T cell exhaustion.23 Therefore, we were interested in measuring the duration of the stimulatory signal that our aAPC-ΔLDLR provide. To study the kinetics of T cell activation and irradiated aAPC disappearance from the coculture during CAR-T production, T cells and irradiated aAPC-ΔLDLR cocultures were set up at a 2:1 ratio. Large T cell-aAPC-ΔLDLR clusters were detected on microscopy 18 hours after initiation of the coculture, demonstrating T cell activation at this early timepoint (figure 1E). The number of aAPC-ΔLDLR was determined daily by total cell count and flow cytometry. The number of irradiated aAPC-ΔLDLR in the coculture started declining by day 1 and reached 0 by day 3 (figure 1F). Importantly, we also demonstrate that by the time of harvest of a CAR-T product, no residual aAPC remained.
aAPC-ΔLDLR efficiently expand CAR-T cells
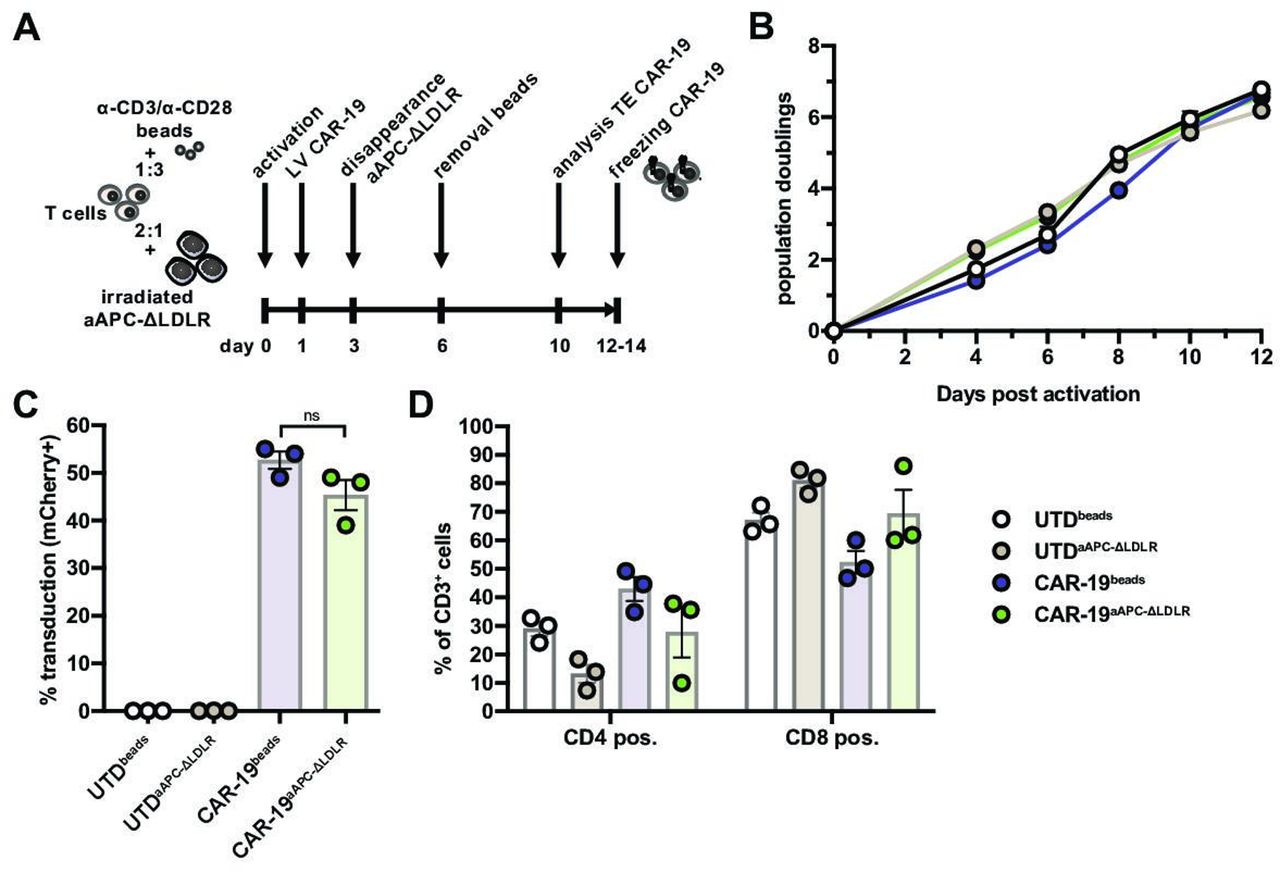
Having established that our aAPC-ΔLDLR cell line is resistant to VSV-LV transduction, we proceeded to test their capacity to stimulate expansion of CAR-T cells in comparison to the standard method using beads coated with anti-CD3 and anti-CD28 antibodies (“beads”). In the current study, we used a second-generation anti-CD19 CAR with a CD8 transmembrane domain and 4-1BB and CD3ζ intracellular domains, reflecting one of the clinically approved constructs. To facilitate measurement of transduction efficiency, the vector contained a second transgene coding for the fluorescent reporter mCherry. T cell:aAPC-ΔLDLR cocultures were set up at a 2:1 ratio, while T cell:beads cocultures were set up at a 1:3 ratio. On day 1 post activation, lentiviral vector encoding the CD19 CAR was added to the cultures at an MOI of 5. In the appropriate cultures, beads were removed on day 6 of the coculture (figure 2A). On day 10, CAR transduction efficiency of CD3+ cells was measured based on mCherry expression by flow cytometry. In parallel, donor-matched T cells that had been activated with either aAPC-ΔLDLR or beads but not transduced (UTD) were cultured as controls. During 14 days of ex vivo culture, the expansion kinetics and CAR-19 transduction efficiency of primary human T cells (CAR-19) were comparable between aAPC-ΔLDLR-based (CAR-19aAPC-ΔLDLR) and bead-based (CAR-19beads) activation (figure 2B,C). Looking at CD4+/CD8+ ratios, aAPC-ΔLDLR trended toward preferentially expanding CD8+ over CD4+ T cells (figure 2D), as compared with beads.

Artificial antigen-presenting cells (aAPC)-Δ low-density lipoprotein receptor (LDLR) efficiently expand chimeric antigen receptor (CAR)-19 T cells. (A) Schematic illustrating the timeline for CAR-T production comparing aAPC-ΔLDLR- and bead-based T cell activation. (B) Expansion kinetics and (C) CAR-19 transduction efficiency of aAPC-ΔLDLR- and bead-stimulated primary human T cells during 14 days of ex vivo culture. (D) CD4+/CD8+ ratios of UTD and CAR-19 T cells at the end of the expansion. Data points/bars depict mean ± SEM of 3 normal donors. Unpaired, two-tailed t-test, ns = p ≥ 0.05.
aAPC-ΔLDLR-expanded CAR-19 T cells show potent in vitro effector function against target cells
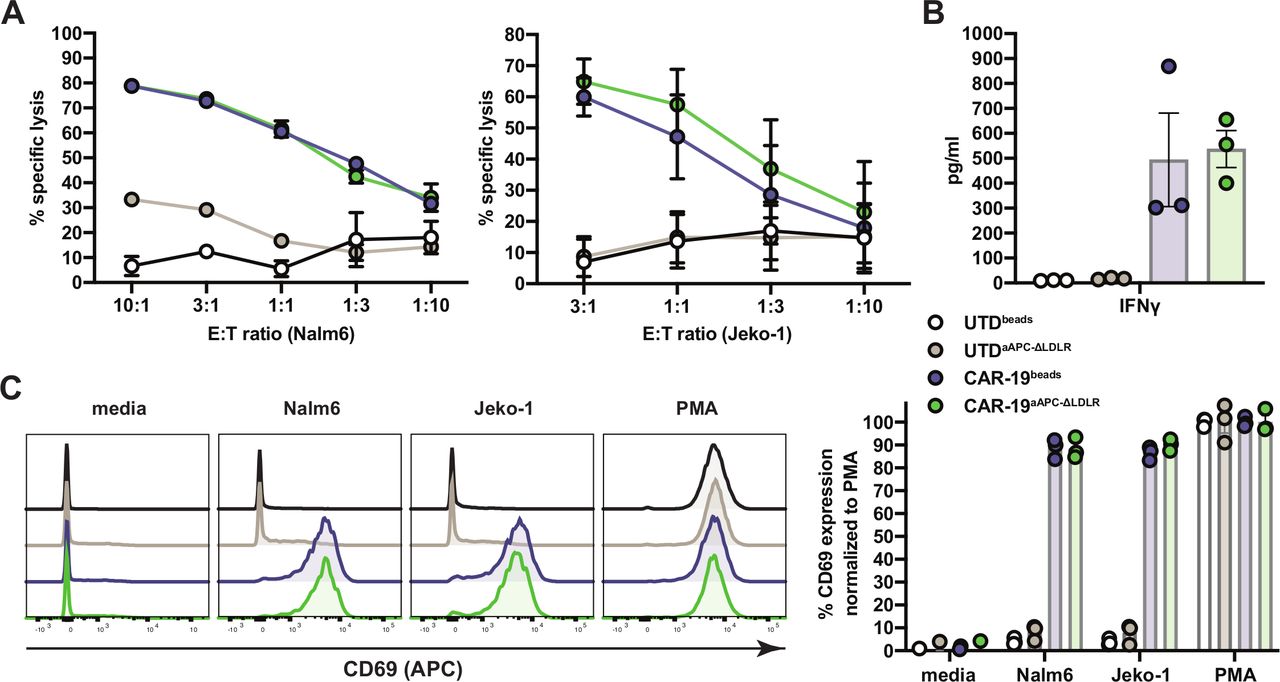
We proceeded to test the potency of CAR-19aAPC-ΔLDLR to induce antitumor immune responses in vitro. Cocultures of UTD and CAR-19 T cells with Nalm6 and Jeko-1 target cells were set up at various E:T ratios. Cytotoxicity was measured in a luciferase-based assay. Nalm6 and Jeko-1 target cells were lysed at high efficiency by CAR-19aAPC-ΔLDLR and CAR-19beads T cells (figure 3A). Analysis of supernatants from coculture of CAR-19 T cells with Nalm6 targets showed antigen-specific production of interferon gamma at comparable levels for CAR-19aAPC-ΔLDLR and CAR-19beads T cells (figure 3B). To further assess antigen-mediated T cell activation, we measured upregulation of the surface marker CD69 after an overnight stimulation with Nalm6 and Jeko-1 target cells. CAR-19aAPC-ΔLDLR and CAR-19beads T cells both showed strong CD69 upregulation when cocultured with target cells compared with baseline and the positive control, phorbol myristate acetate (PMA) (figure 3C).

Artificial antigen-presenting cells (aAPC)-Δ low-density lipoprotein receptor (LDLR)-expanded chimeric antigen receptor (CAR)-19 T cells display potent in vitro effector function against target cells. (A) Cocultures of UTDbeads, UTDaAPC-ΔLDLR, CAR-19beads, and CAR-19aAPC-ΔLDLR T cells with Nalm6 and Jeko-1 target cells were set up at various effector:target (E:T) ratios, as indicated on the x-axis, and cytotoxicity was measured in a luciferase-based assay. (B) Analysis of interferon γ (IFNγ) production in supernatants of UTDbeads, UTDaAPC-ΔLDLR, CAR-19beads, and CAR-19aAPC-ΔLDLR T cells after 8 hours coculture with human Nalm6 ALL cells at a 1:1 E:T ratio. Cytokines were measured by Luminex assay in technical duplicates. (C) Activation of UTDbeads, UTDaAPC-ΔLDLR, CAR-19beads, and CAR-19aAPC-ΔLDLR T cells after overnight stimulation, with Nalm6 and Jeko-1 target cells at a 1:1 E:T ratio. Surface expression of CD69 was measured on UTD (CD3+) and CAR-19 T cells (CD3+mCherry+) by flow cytometry. Histograms depict a representative sample of three normal donors. Data points/bars indicate mean ± SEM of three normal donors.
aAPC-ΔLDLR-expanded CAR-19 clear high-tumor burden ALL in a xenograft mouse model
Next, we compared the in vivo antitumor efficiency of CAR-19 T cells produced using either bead-based or aAPC-ΔLDLR-based expansion in a xenograft mouse model of ALL. NSG mice received an intravenous injection of 1×106 luciferized Nalm6 leukemia cells. Engraftment was confirmed by BLI 1 day before T-cell injection. On day 0, mice were injected with a single intravenous dose of normalized CAR19 T cells or UTDs that had been aAPC-ΔLDLR expanded or bead expanded. Tumor burden was monitored twice weekly by BLI. CAR-19 T cell persistence was measured in peripheral blood weekly by flow cytometry, as well as in the bone marrow and spleen at the end of the experiment (figure 4A). In both UTD-treated groups, tumor burden progressed rapidly. By day 15 post T cell administration, these mice had died or needed to be euthanized. In contrast, both CAR-19-treated groups cleared the high tumor burdens and were all still alive at the end of the experiment, except for one likely unrelated death (figure 4B–D). In the peripheral blood, CAR-19beads and CAR-19aAPC-ΔLDLR showed different expansion kinetics with cell numbers peaking on days 14 and 18 after CAR-T administration, respectively (figure 4E). However, because of donor variability, these differences did not reach statistical significance at any of the analyzed timepoints. In addition, our in vivo model was limited to weekly testing for CAR-19 persistence in the peripheral blood, which, in the case of this aggressive ALL model, did not allow for continuous monitoring of CAR-19 persistence. At the end of the experiment, persistence of CAR-19 T cells in the bone marrow and spleen was comparable for both manufacturing methods (figure 4F).
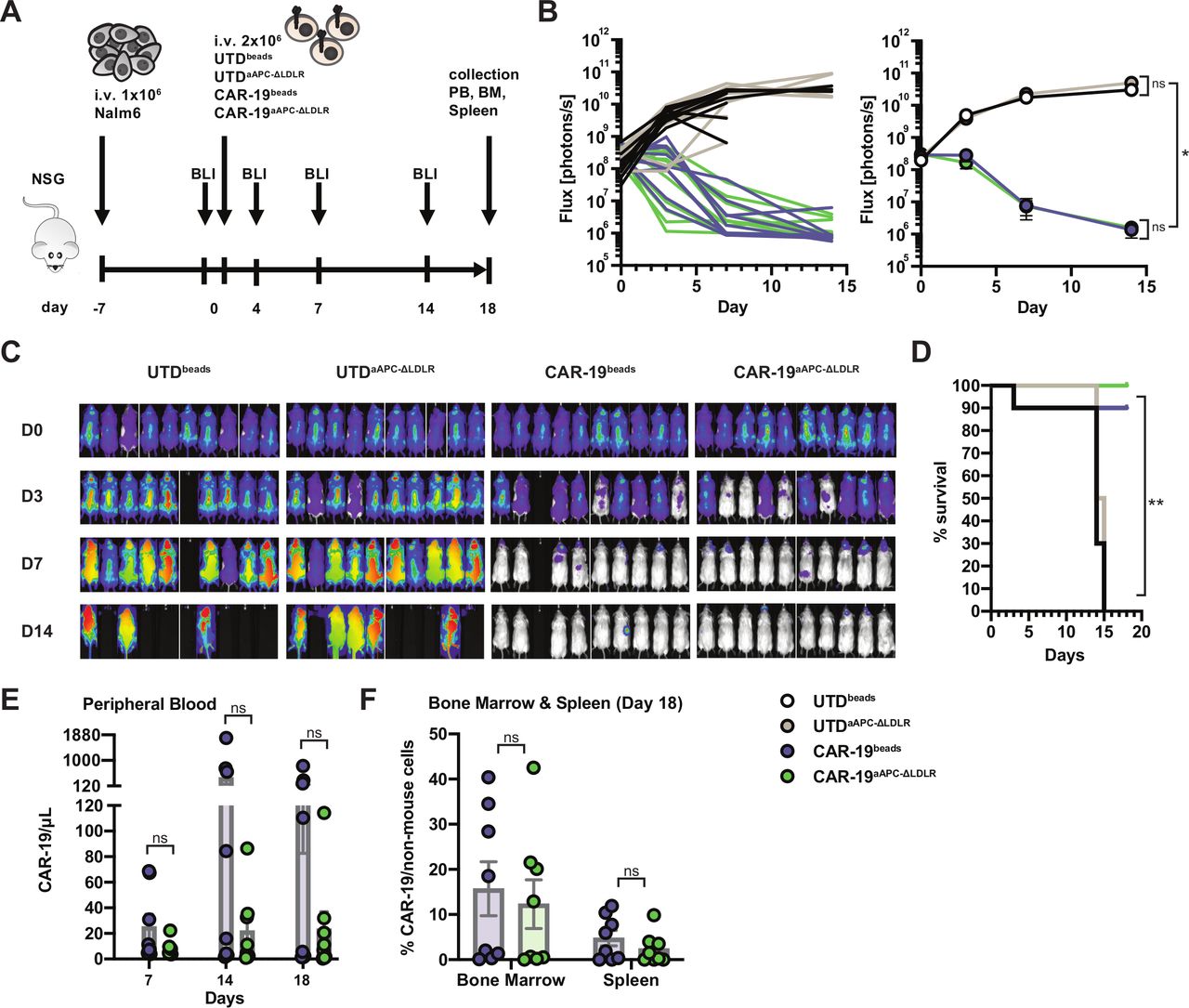
Artificial antigen-presenting cells (aAPC)-Δ low-density lipoprotein receptor (LDLR)-expanded chimeric antigen receptor (CAR)-19 T cells specifically eradicate ALL in vivo. (A) Experimental design to assess antitumor efficiency of UTDbeads, UTDaAPC-ΔLDLR, CAR-19beads and CAR-19aAPC-ΔLDLR T cells in a xenograft model of ALL. NSG mice were engrafted with 1×106 Nalm6 tumor cells, and their clinical condition as well as tumor burden, measured by bioluminescence imaging (BLI), were monitored closely. One week after tumor injection, the mice were randomized to receive a single dose of either UTDbeads, UTDaAPC-ΔLDLR, CAR-19beads, or CAR-19aAPC-ΔLDLR T cells normalized to the same number of total T cells and transduction efficiency. (B) Quantification of flux (photons/s) of ALL xenografts for each individual mouse (left) and for the four experimental groups (right) over time. Two-way analysis of variance test, ns=p > 0.5, *=p≤ 0.05. (C) Representative BLI of ALL xenografts in the four experimental groups over time. (D) Survival curves for each group using Kaplan-Meier product-limit estimation. Log-rank test, **=p ≤ 0.001 (E) CAR-19 T cell (CD3+mCherry+) persistence measured in the peripheral blood as well as (F) bone marrow and spleen at the indicated time points by flow cytometry. Data points show mean ± SEM of two normal donors with five mice per group. Unpaired, two-tailed t-test, ns=p > 0.5.
aAPC-ΔLDLR efficiently expand CAR-19 T cells in the presence of contaminating monocytes and from unmanipulated starting material
Monocyte contamination of the starting T cell source inhibits bead-based CAR-T expansion and, in some cases, leads to production failure.7 This can be due either to inadvertent transduction and “sink” of the vector, or due to monocyte phagocytosis of the beads and therefore of the T-cell-stimulating molecules. Therefore, we investigated whether our aAPC-ΔLDLR-based approach for expanding CAR-T is affected by the presence of monocytes in the starting material. Using either purified T cells or PBMC as T cell source, we compared the CAR-19 T cell yield with bead-based and aAPC-ΔLDLR-based activation. The PBMC starting source on average contained 11% (5%–17%) monocytes after thawing, while there were no monocytes detected in the purified T cell samples (figure 5A). Expansion kinetics and CAR transduction efficiency were comparable for both activation methods when using purified T cells, resulting in similar CAR-19 yields. In contrast, when using PBMC as starting material, CAR-19 transduction efficiency was twofold higher via aAPC-ΔLDLR activation compared with bead activation (p=0.02) and resulted in a twofold higher CAR-19 yield (p=0.08; figure 5B–D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Artificial antigen-presenting cells (aAPC)-Δ low-density lipoprotein receptor (LDLR) efficiently expand chimeric antigen receptor (CAR)-19 T cells in the presence of contaminating monocytes and from complex starting material. (A) Percentage of contaminating monocytes (CD14+) in matched PBMC and purified T cell samples from three normal donors after cryopreservation. The mean percentage is indicated at the top of each bar graph. (B–D) CAR-19 transduction efficiency, population doublings and CAR-19 yield of aAPC-ΔLDLR- and bead-stimulated primary human-purified T cells and PBMC during 12 days of ex vivo culture. Data points/bars depict mean ± SEM of three normal donors. Unpaired, two-tailed t-test, ns = p ≥ 0.05, * = p < 0.05. (E) Cellular composition of fresh whole leukopak samples from two normal donors. Percentages refer to total live cells. (F)–(H) CAR-19 transduction efficiency, lymphocyte population doubling and CAR-19 yield of aAPC-ΔLDLR- and bead-stimulated fresh whole leukopak cells during 12 days of ex vivo culture. Data points and mean of two normal donors depicted.
Since monocyte contamination of the starting T cell source did not impede aAPC-ΔLDLR-based CAR-19 manufacturing, we reasoned that aAPC-ΔLDLR may also be capable of efficiently expanding CAR-T from unmanipulated starting material, like whole leukapheresis products (figure 5E). Using both activation methods, we observed comparable lymphocyte population doublings from whole unmanipulated leukopaks that were used fresh. However, taking the transduction efficiency into consideration, aAPC-ΔLDLR activation resulted in more efficient expansion of CAR-19 T cells from whole unmanipulated leukopaks, with 50% higher CAR-19 T cell yield than bead activation (figure 5F–H).
Discussion
CAR-T therapy has become a standard treatment for patients with certain aggressive B cell malignancies and is currently being investigated for the treatment of multiple other cancers. Even so, the high cost of CAR-T cell manufacturing represents a major barrier to a broader clinical application of CAR-T therapies.24 Among the key factors contributing to manufacturing challenges are the limited production of clinical-grade viral vectors, the single-use GMP-quality reagents, and the variability in composition of the starting cellular material, which pose a challenge to reproducibly obtaining cell products that meet the rigorous release criteria.25 26 Therefore, strategies aiming to maximize utilization of the viral vector, minimize the need for single-use reagents, and reduce the risk of manufacturing failure are likely to improve the success rate and reduce the cost of engineered T cell production for clinical applications.
In this study, we developed K562-based aAPC that have genetically encoded T cell stimulation and are resistant to VSV-LV transduction (aAPC-ΔLDLR). We demonstrated that aAPC-ΔLDLR efficiently expand CAR19 T cells with high antitumor potency in preclinical models of ALL and B-cell lymphoma. Furthermore, in contrast to standard bead-based activation, our strategy for T cell activation resulted in robust CAR-T expansion even from unpurified starting material like PBMC or unmanipulated whole leukopaks.
Cell-based aAPC are an attractive strategy to expand T cells for adoptive transfer because they are customizable, cost-efficient, and form more natural immune synapses than bead-based aAPC.15 16 However, one major drawback of using cell-based aAPC for expansion of lentivirally engineered T cells is that the cell-based aAPC compete with the T cells for the viral vector while in coculture. Therefore, we generated K562-based aAPC that are resistant to incidental lentiviral transduction by deletion of the LDLR. Compared with previous K562-based aAPC for T cell expansion, our aAPC-ΔLDLR allow for a tighter regulation of the transduction processes. This is especially important for clinical scale CAR-T manufacturing, where fine control over the viral titer and transduction process is essential in order to assure consistency between manufacturing runs for different patients.
Current methods of clinical CAR-T manufacturing use antibody-coated beads for T cell stimulation that are usually removed from the culture 9–11 days post activation.8 A prolonged stimulation with sustained TCR signaling can cause exhaustion of T cells and reduce their proliferative and cytotoxic capacity.23 In contrast, transient stimulation has been shown to result in T cell products with favorable phenotypic and functional properties. Importantly, K562-based aAPC expressing CD3/CD28 counter receptors have been shown to promote maintenance of a stem cell-like memory T cell phenotype compared with anti-CD3/anti-CD28-coated beads.27 28 In our study, we found that irradiated aAPC-ΔLDLR provided stimulation to T cells for less than 3 days post activation. An additional advantage of using irradiated cell-based over bead-based aAPC is that no additional bead removal step is required, making the manufacturing process easier.
During the manufacturing process, contaminating monocytes in the cell therapy starting material can inhibit CAR-T cell expansion and even lead to manufacturing failure.7 Patients with cancer have elevated numbers of circulating myeloid-derived suppressor cells, with monocyte-like and granulocyte-like phenotypes,29 30 which may interfere with the manufacturing process. Using bead-based activation of PBMC and whole unmanipulated leukopak to manufacture CAR-T cells, we obtained a lower yield of CAR-T cells compared with starting with purified T cells, confirming the negative impact of contaminating cell populations on CAR-T expansion. In contrast, when using aAPC-ΔLDLR activation, we did not observe a difference in CAR-T expansion in the presence of contaminating cell populations. The negative impact of contaminating cell populations on CAR-T expansion in a clinical setting may be even more significant than our study found, since we performed all experiments with healthy blood donor cells. Additional studies would also be required to test our aAPC-ΔLDLR in the context of large-scale CAR-T manufacturing using a bioreactor system.
Use of aAPC-ΔLDLR is an attractive approach for CAR-T manufacturing that generates CAR-T cells that mount potent in vitro and in vivo antitumor immune responses. Generation of lentivirally transduced T cells using aAPC-ΔLDLR may offer a simpler and more cost-efficient manufacturing procedure than currently available methods.
Acknowledgments
The authors thank Trisha R. Berger for assistance with editing the manuscript.
References
Footnotes
Twitter @SchmidtsAndrea, @BryanDChoi, @MJFzeta, @BKleinstiver, @JKeithJoung, @MarcelaMaus
Contributors AS and MVM designed the study. AS, LCM, AAS, AAB, ACB, IS, RCL, FB, MSP, BPK, JKJ and MVM designed and executed individual experiments. AS, LCM, BC, MJF, SRB, MBL and MVM analyzed and interpreted data. All authors read and approved the final manuscript.
Funding AS received support from the German Cancer Aid Mildred-Scheel Post-Doctoral Research Fellowship. This work and MVM was supported by NIH 1R01CA238268. JKJ and colleagues were supported by an NIH Maximizing Investigators’ Research Award (MIRA; R35 GM118158). MBL received support by NIH 2T32CA071345-21A1. BPK was supported by NIH R00-CA218870. IS is a Lymphoma Research Foundation Grantee. SRB received support from NIH 5T32CA009216-39.
Competing interests Some of the authors have filed provisional patent applications related to the use of the aAPC’s described herein. BPK is an inventor on various patents and patent applications that describe gene editing and epigenetic editing technologies. BPK is a consultant for Avectas and an advisor for Acrigen Biosciences. JKJ has financial interests in Beam Therapeutics, Editas Medicine, Excelsior Genomics, Pairwise Plants, Poseida Therapeutics, Transposagen Biopharmaceuticals, and Verve Therapeutics (f/k/a Endcadia). JKJ’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. JKJ is a member of the Board of Directors of the American Society of Gene and Cell Therapy. JKJ is a coinventor on various patents and patent applications that describe gene editing and epigenetic editing technologies.
Patient consent for publication Not required.
Ethics approval All experiments in this study were performed under protocols reviewed by the Massachusetts General Hospital Institutional Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author note ACB is currently at ArsenalBio. and FB is currently at Tmunity Therapeutics Inc.