Article Text

Abstract
Checkpoint blockade immunotherapy (CBT) can induce long-term clinical benefits in patients with advanced cancer; however, response rates to CBT vary by cancer type. Cancers of the skin, lung, and kidney are largely responsive to CBT, while cancers of the pancreas, ovary, breast, and metastatic lesions to the liver respond poorly. The impact of tissue-resident immune cells on antitumor immunity is an emerging area of investigation. Recent evidence indicates that antitumor immune responses and efficacy of CBT depend on the tissue site of the tumor lesion. As myeloid cells are predominantly tissue-resident and can shape tumor-reactive T cell responses, it is conceivable that tissue-specific differences in their function underlie the tissue-site-dependent variability in CBT responses. Understanding the roles of tissue-specific myeloid cells in antitumor immunity can open new avenues for treatment design. In this review, we discuss the roles of tissue-specific antigen-presenting cells (APCs) in governing antitumor immune responses, with a particular focus on the contributions of tissue-specific dendritic cells. Using the framework of the Cancer-Immunity Cycle, we examine the contributions of tissue-specific APC in CBT-sensitive and CBT-resistant carcinomas, highlight how these cells can be therapeutically modulated, and identify gaps in knowledge that remain to be addressed.
- antigen presentation
- dendritic cells
- immune evation
- tumor microenvironment
- inflammation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Now approved for over 11 cancer indications, checkpoint blockade immunotherapy (CBT) can induce durable antitumor immunity in patients with advanced cancer.1 However, CBT efficacy varies by cancer type. Among cancers originating in non-lymphoid tissues, CBT achieves best results against malignant melanoma1 and lung2 and kidney3 carcinomas. However, for other carcinomas, including pancreatic cancer,4 non-virally induced liver cancer,5 ovarian cancer,6 7 and breast cancer,8 9 the fraction of patients that benefit from CBT is dishearteningly low. Understanding how to extend the benefits of this therapy to a larger number of patients is of great therapeutic interest.
Several factors influence the sensitivity of different tumors to CBT. Tumor-intrinsic factors, such as mutational load, oncogenic signaling pathways, and antigen presentation ability, undoubtedly impact disease progression and treatment outcomes.10 However, tumor-extrinsic factors, such as tissue microenvironment and composition of tissue-resident immune cells, can also shape antitumor immune responses and sensitivity to CBT. Indeed, studies suggest that antitumor immune responses against melanoma and non-small-cell lung cancer (NSCLC) vary by tissue site of metastasis.11 12 Moreover, colorectal and ovarian cancer case reports describe interlesion differences in immune infiltration.13 14 Within a single patient, non-responding lesions can evade immune control by distinct mechanisms, including exclusion or dysfunction (exhaustion) of cytotoxic T cells.14 Given that myeloid cells can impact antitumor immunity15–18 and the observed intertissue diversity of these cells19–21 (tables 1 and 2), it is conceivable that tissue-specific myeloid antigen-presenting cells (APCs) play an important role in controlling local responses to tumors. Comprising dendritic cells (DCs), macrophages, and monocytes, myeloid cells can directly influence T cell phenotype and function, and ultimately promote or suppress antitumor immunity.22 Therefore, it is critical to understand the composition of tissue-resident myeloid cells, as they can differentially impact tissue site responses to CBT.
Murine DC and macrophage subsets and surface markers in different tissues
Human DC and macrophage subsets and surface markers in different tissues
In this review, we examine the characteristics and functions of tissue-resident myeloid APC in different tumor types with specific emphasis on the role of DC in governing the strength of local antitumor immune responses. We start by reviewing the general contributions of myeloid APC to productive antitumor immunity in the Cancer-Immunity Cycle. We then discuss known tissue-specific DC and macrophage subsets and their functions in CBT-sensitive carcinomas, such as lung and kidney, as well as CBT-refractive carcinomas, such as breast, ovary, pancreas, and liver. As the contributions of APC to immune responses against melanoma have been recently covered,23–26 we focus our review on carcinomas that vary in sensitivity to CBT. We also highlight recent advances in our understanding of how these cells can be modulated to enhance tumor control and identify gaps in knowledge that remain to be addressed.
The role of myeloid APC in the Cancer-Immunity Cycle
Our current understanding of the requirements for the induction of antitumor immunity is described in the Cancer-Immunity Cycle27 (figure 1). The cycle is initiated when myeloid APC, sensing various tumor-derived danger signals, infiltrate the tumor microenvironment (TME) and capture tumor antigens. These activated APC then migrate to the draining lymph nodes to prime tumor-reactive T cells, and the cycle concludes with T cell infiltration into the tumor and T cell-mediated tumor destruction.
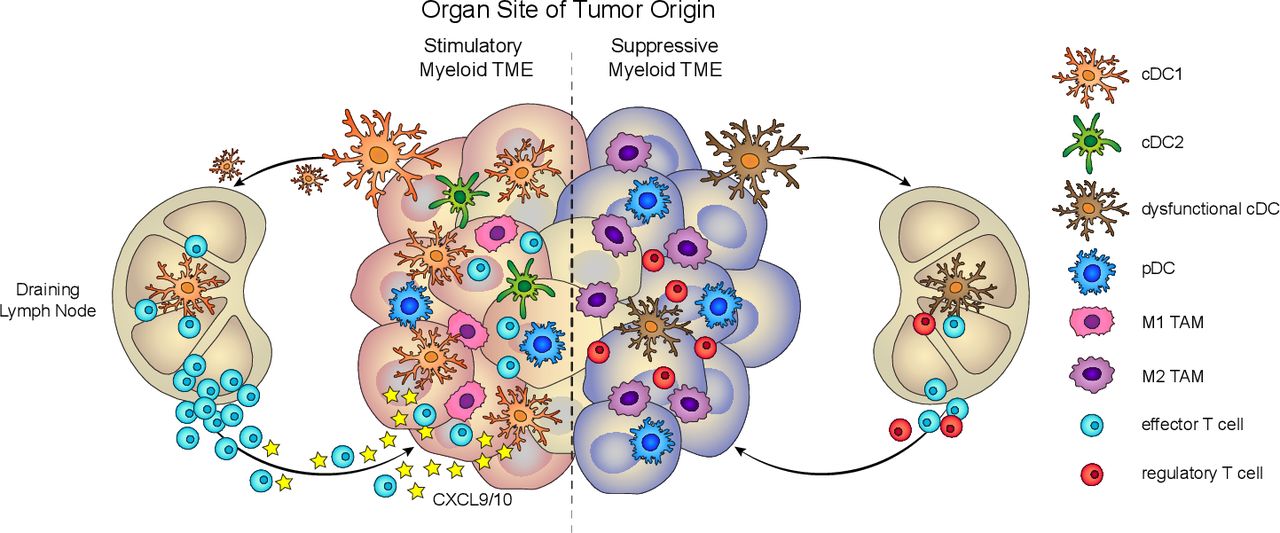
Tissue-specific myeloid cell composition impacts antitumor immunity. Left, Productive antitumor immunity depends on the presence of stimulatory myeloid cells, such as cDC1, in the TME. cDC1 contributes to several critical functions, such as cross-presentation, antigen transport to the lymph node, T cell priming, and T cell recruitment, to drive tumor-reactive T cell responses. Right, Dysfunctional antitumor immunity can arise from tumors that are predominantly infiltrated by suppressive myeloid cells, such as M2 TAM and functionally impaired DC. cDC, conventional DC; DC, dendritic cell; pDC, plasmacytoid DC; TAM, tumor-associated macrophage; TME, tumor microenvironment.
While monocytes and macrophages contribute to the Cancer-Immunity Cycle, early evidence established a role for myeloid DC as key initiators of immunity through their ability to transport antigens from the periphery to lymphoid organs.28 The DC compartment is classified into two main lineages: myeloid or conventional DC, which comprises cDC1 and cDC2 subsets, and plasmacytoid DC (pDC).19 29–31 However, the DC compartment is further diversified by the inclusion of monocyte-derived DC (moDC) subsets, such as the tumor necrosis factor-α (TNF-α)/inducible nitric oxide synthase (iNOS)-producing TiP-DC,32 33 and by the recent identification of AXL+ Siglec-6+ (AS)-DC and multiple pre-DC and cDC2 subtypes.34–36 As the predominant cross-presenting DC subset, Batf3-driven CD8α+/CD103+ cDC1 are considered to be the key mediators of a T cell-inflamed TME, carrying out canonical roles of antigen transport and cross- priming.37–40 Novel functions for cDC1 have also been recently reported. Tumor-residing cDC1 can secrete CXCL9 and CXCL10 chemokines to recruit effector T cells into the tumor,16 and they can locally restimulate effector T cells in the TME.15 While the roles of other DC subsets in antitumor immunity are less well-defined, recent work suggests that they can also contribute to antitumor immunity. cDC2 are critical for stimulating tumor-reactive effector CD4+ T cell responses,41 42 and pDC can enhance antitumor immune responses via production of large amounts of type-I-interferons (type-I-IFNs).43
The Cancer-Immunity Cycle, however, depicts an oversimplified view of productive antitumor immunity. It does not factor in the tissue microenvironment, such as tissue-specific myeloid cells, which can skew antitumor immune responses (figure 1) and impact CBT efficacy. Supporting this notion, the environmental context of the tissue can induce tissue-specific DC and macrophages that dampen antitumor immune responses and promote an immunosuppressive TME.44 45 Known mechanisms for tumor tissue-induced rewiring of APC responses include tumor-intrinsic signaling pathways, such as IFNγ, MAPK, Wnt/β-catenin, and COX-2, which can directly compromise DC function or lead to DC exclusion from the TME, thereby enabling immune escape and impairing CBT efficacy.18 46–50 In the absence of tumor, healthy skin tissue can condition DC to express an IFNγ-dependent homeostatic program that is enriched in tolerance-related genes.50 51 This microenvironment-dependent plasticity of the myeloid compartment highlights the importance of considering the contribution of tissue-specific APC to antitumor immune responses.
Understanding the functional nuances of tissue-specific APC will inform development of novel cancer immunotherapies. Several DC-based vaccination approaches are currently being developed to restore functional DC responses in the contexts of cancer and viral infections.52 These DC-targeted therapies have the potential to enhance responses to CBT, given that DC are directly involved in the molecular interactions targeted by CBT: DC can suppress T cell function via CTLA-4 engagement,53 and recent work has identified DC-specific Programmed Death Ligand 1 (PD-L1) as a critical regulator of T cell-driven immunity.54 Broader strategies to directly modulate the tumor myeloid compartment provide additional therapeutic opportunities.55 56 Thus, dissecting the contributions of tissue-specific myeloid cells to antitumor immunity will enlighten our understanding of tumor type-specific sensitivity to CBT and enable us to harness the potential of these cells in new therapies.
Myeloid APC in CBT-sensitive carcinomas
Non-small cell lung cancer
NSCLC responds to anti-PD-1 (nivolumab) and anti-CTLA-4 (ipilimumab) combination CBT, with an objective response rate of 33%.2 Given that the most common cause of lung cancer is tobacco smoke, it is thought that the resulting high mutational burden is the main driver of response.57 However, while tumor-intrinsic characteristics such as mutational load can impact lung cancer progression and immune detection, activated APC need to be present in the tissue to initiate and facilitate a productive antitumor response (reviewed in the study by Kopf et al 58).
In the context of respiratory virus infection, the stimulatory capacity of lung DC subsets can induce protective immunity. Lung cDC1 effectively cross-present viral antigens, transport antigens to the draining lymph node, activate CD8+ T cells, induce their trafficking to the infection site, and promote prolonged survival of T cells in the lung tissue.59–64 Interestingly, at baseline lung cDC1 are considerably more long-lived than their counterparts in other non-lymphoid tissues.20 Later in the course of infection, lung cDC2 become the dominant CD8+ T cell-stimulating subset, inducing central memory and resident memory cells.63 65 66 A more granular view of the cDC2 compartment suggests that it comprises a mixture of subsets, including the traditional cDC2, which can migrate to the lymph node and preferentially stimulate CD4+ T cells, and a novel inflammatory cDC2 population (inf-cDC2), which appears in the context of respiratory viral infection and allergy.67 Intriguingly, inf-cDC2 can acquire cDC1 characteristics in a type-I-IFN-dependent manner and become capable of optimally priming CD4+ and CD8+ T cells.67 The infected lung also contains moDC, which are non-migratory and are poor activators of CD4+ or CD8+ T cells, compared with cDC.67 Additionally, lung pDC can produce large amounts of type-I-IFN in response to infection with pneumonia virus of mice, inducing a proinflammatory antiviral response.68
Although the immune-stimulatory potential of lung DC can have protective effects in cancer, the lung TME inhibits their antitumor functions (figure 2), and efforts are underway to test the efficacy of combining a DC-based vaccine with CBT.69 The protective role of cDC1 is clearly illustrated in studies of mouse models of lung adenocarcinoma: increasing lung cDC1 number results in a reduced tumor burden, while deleting them causes a lack of CD8+ T cell infiltration and increased tumor progression.70 71 The presence of a cDC1 gene signature is also associated with better outcomes in patients with lung adenocarcinoma.15 48 However, human lung tumors contain a reduced proportion of cDC1 compared with healthy tissues, suggesting tumor exclusion.72 In addition to affecting DC numbers, lung tumors in both mice and humans can also modulate DC-stimulatory capacity, by inducing a regulatory phenotype characterized by expression of T-helper 2 (TH2) response genes.70 These regulatory DC are capable of both inducing differentiation of naive CD4+ T cells into FoxP3+ regulatory T (Treg) cells and activating antigen-specific CD8+ T cells.70 Intriguingly, administration of interleukin 4 (IL-4) blocking antibodies to lung tumor-bearing mice reprograms these DC toward a more stimulatory phenotype, characterized by increased IL-12 production and enhanced ability to activate naive antigen-specific CD8+ and CD4+ T cells.70 Further research is needed to determine whether this regulatory DC subset is related to the ‘activated’ CCR7+ DC subset previously observed in mouse and human lung adenocarcinoma samples73 and/or the inf-cDC2 characterized in the virally infected lung.67 Furthermore, pDC become more abundant and immunosuppressive in human lung tumors compared with healthy tissue,74 and pDC ablation can improve responses to TLR9-agonist therapy in mouse models of lung cancer.75 Importantly, single-cell RNA-sequencing analysis of mouse and human lung adenocarcinoma samples suggests that DC subsets are highly conserved between mice and humans,73 highlighting the relevance of using preclinical models in the study of lung cancer.

{kind=link}
{kind=link}
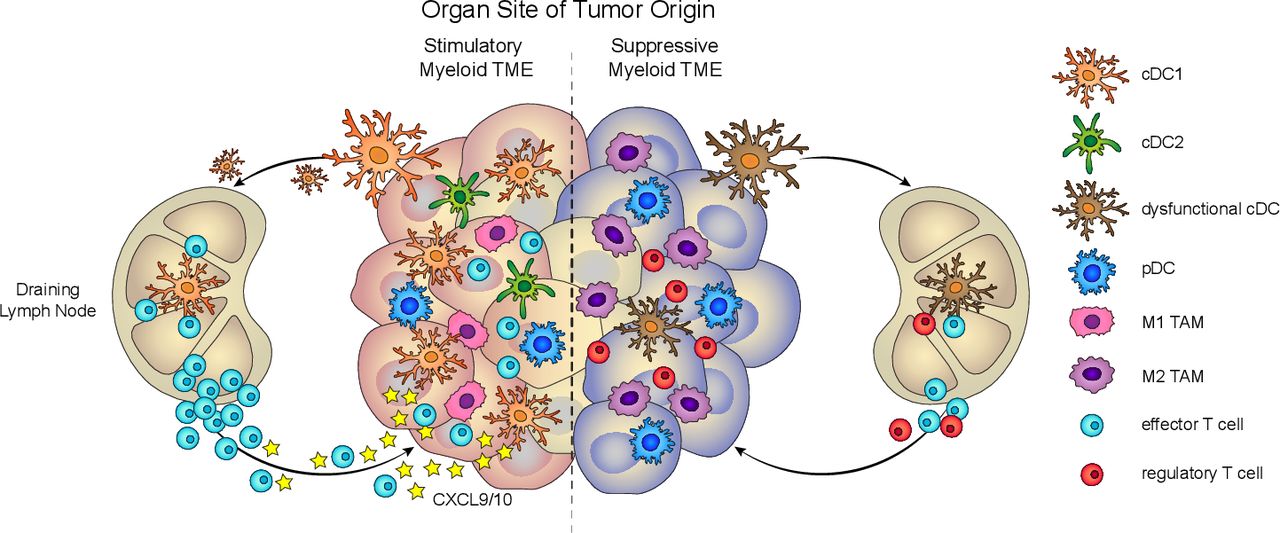
The myeloid immune microenvironment varies depending on the tissue site of tumor origin. CBT-resistant carcinomas (pancreas, liver, ovary, and breast) are generally heavily infiltrated by suppressive myeloid cells, such as TAM and tolerogenic DC subsets, while stimulatory cDC1 are scarce and prone to becoming dysfunctional. CBT-sensitive carcinomas (lung, kidney) similarly harbor suppressive TAM and inhibitory DC, but they also contain protective stimulatory cDC1. The balance of immune-potentiating and immunosuppressive myeloid cells at each tissue site impacts antitumor immunity responses and sensitivity to CBT. CBT, checkpoint blockade immunotherapy; cDC, conventional DC; DC, dendritic cell; moDC, monocyte-derived DC; pDC, plasmacytoid DC; TAM, tumor-associated macrophage.
In contrast to DC, the role of lung macrophages in lung cancer is less understood. Alveolar macrophages (AM) comprise 90%–95% of cellular content in the air space of alveoli and can perform both tolerogenic and inflammatory functions.58 However, unlike cDC, AM are ineffective at activating T cells directly due to their low expression of costimulatory molecules.76 Interestingly, on exposure to bacteria, AM can rapidly phagocytose them and traffic to the lymph node.77 The significance of this is unclear, but an intriguing possibility might be that AM transfer foreign antigens to lymph node-resident DC. In the context of cancer, lung macrophages are suggested to be skewed to the protumorigenic M2-like phenotype (figure 2).73 Beyond AM, multiple other macrophage subsets were identified in lung adenocarcinoma, some of which appear to be species-specific.73 The roles of these distinct macrophage subsets in lung cancer remain to be elucidated.
Renal cell carcinoma
Renal cell carcinoma (RCC) is highly responsive to anti-PD-1 (nivolumab) and anti-CTLA-4 (ipilimumab) combination CBT, with an objective response rate of 42%.3 Compared with other cancers types, RCC has the highest number of insertion/deletion mutations, which are considered immunogenic78 and correlate with sensitivity to anti-PD-1 therapy in patients with advanced RCC.79 The responsiveness of RCC to CBT implies that renal APC must be largely stimulatory. However, as a filtration organ, the kidney concentrates a large number of antigens and must maintain a careful balance between tolerance and immunity.
Renal DC appear to have both proinflammatory and anti-inflammatory roles, which are context specific (recently reviewed in the study by Kurts et al 80). Among resident DC subsets, renal CD103+ cDC1 are a minor population that exhibits the maximal antigen presentation capacity.81 At baseline, these cells express high levels of CCR7 and CD86, can stimulate CD8+ T cells, and induce differentiation of T-helper 1 (TH1) cells.81 82 At the same time, renal cDC1 act to prevent excess inflammation in the tissue by maintaining IL-10-producing Treg cells83 and inhibiting neutrophil recruitment.84 The dominant renal DC subset is CD11b+ cDC2, which express high levels of Major Histocompartibility Complex Class-II (MHC-II) and C-C Chemokine Receptor Type 7 (CCR7), preferentially prime CD4+ T cells and can induce a high proportion of Treg cells.81 82 84 These cells can be proinflammatory, as a large number of cDC2 appear to migrate to the lymph node during systemic LPS administration,81 and cDC2 can recruit neutrophils during nephrotoxin serum-induced kidney injury.84 pDC were not detected in the mouse kidney.81 85 In the human kidney, cDC1, cDC2, and pDC were identified.86
Although the presence of the DC signature in tumors correlates with sensitivity to CBT in patients with kidney cancer,87 the RCC TME can skew renal myeloid DC and pDC toward an immature state rendering them incapable of migrating to the draining lymph node88 (figure 2). In culture, RCC cells release vascular endothelial growth factor (VEGF) and IL-6, which can inhibit the ability of DC to stimulate T cells.89 Furthermore, treatment with RCC-conditioned media can cause DC to downregulate expression of costimulatory molecules and production of IL12p70 and upregulate secretion of TGF-β and IL-10.90 Interstitial immature CD209+ DC from tumor samples of patients with clear cell RCC (ccRCC) secrete high levels of matrix metalloproteinase 9, reduce TH1 cell recruitment, and their frequency correlates with advanced tumor stages.91 Further studies are needed to clarify the roles of distinct DC subsets in kidney cancer.
Macrophages in the RCC TME are abundant, highly heterogeneous and largely protumorigenic (figure 2). Mass cytometry analysis of human ccRCC samples revealed 17 major tumor-associated macrophage (TAM) populations, among which the CD38+CD204+CD206− subset correlated with immunosuppression.92 Mechanistically, TAM can secrete IL-23, which causes Treg cell proliferation, and thus promotes ccRCC tumor immune evasion.93 IL-23 blockade results in prolonged survival of tumor-bearing mice and might be a promising therapeutic target in RCC.93 ccRCC induces M2-polarization of kidney macrophages, causing them to convert T cells to an immunosuppressive phenotype, characterized by expression of PD-1 and TIM-3, and production of IL-10.94 The presence of M2-associated transcripts in tumor samples was associated with reduced ccRCC survival.94
Myeloid APC in CBT-resistant carcinomas
Invasive breast carcinoma
The responsiveness of invasive breast carcinoma to CBT is generally low and varies by subtype.8 Triple-negative breast cancer (TNBC) has thus far demonstrated the most promising outcomes with CBT.8 In fact, the combination of anti-PD-L1 (atezolizumab) with chemotherapeutic nab-paclitaxel was recently approved for treating metastatic TNBC with an objective response rate of 39%,9 whereas anti-PD-L1 monotherapy only resulted in a 10% objective response rate.8 The more common endocrine receptor (ER)-positive/human epidermal growth factor receptor 2 (HER2)-negative and ER-negative/HER2-positive breast cancer subtypes are less responsive to single-agent CBT, and efforts are underway to evaluate various combination therapies.8 9 While the poor response rate is associated with the low mutational load of breast cancers,95 the myeloid cell microenvironment of breast cancer also contributes to the aggressiveness of the disease.96
The contribution of DC subsets in breast cancer is complex, as they have both stimulatory and suppressive roles in the antitumor immune response (figure 2). In a mouse model of spontaneous breast cancer, CD103+ cDC1 and CD11b+ cDC2 subsets exhibited vastly different cytokine and functional profiles.15 cDC1 expressed high levels of the immune-stimulatory cytokine IL-12,15 which is associated with improved chemotherapy responses in human breast cancer.15 97 Compared with other myeloid cell types assessed, cDC1 were found to be superior stimulators of both naive and effector CD8+ T cells.15 In human breast cancers, CD141+ cDC1 could selectively produce type-III-IFNs, which drove a TH1 microenvironment via increased secretion of IL-12p70, IFNγ, CXCL9, and CXCL10.98 Consistent with this finding, a high infiltration of cDC1 in tumors correlated with better outcomes in patients with breast cancer.15 98 However, as cDC1 are a rare cell type in breast cancer,15 their immune-stimulatory contributions are often outweighed by the immune-inhibitory functions of other, more abundant, DC subsets and myeloid cells. In murine breast cancer, CD11b+ cDC2 predominantly produced the immunosuppressive cytokine IL-10 and could only weakly stimulate T cells.15 In another study, a subset of human breast tumor-infiltrating DC that expressed OX40L could drive the development of an inflammatory TH2 microenvironment,99 which is associated with poor prognosis in the clinic.100 Breast tumor-infiltrating pDC exhibited impaired IFNα production and induced the expansion of Treg cells,101 and their presence correlated with poor prognosis in patients.102 Furthermore, human breast cancer cells can express CTLA-4 and suppress DC maturation and function,103 thus contributing to the immunosuppressive environment. A better understanding of the specific functional contributions of DC subsets in breast cancer is needed to identify ways of modulating the tumor immune microenvironment to achieve improved therapeutic outcomes.
Whereas the overall contribution of the DC compartment to antitumor immunity in breast cancer is not clear-cut, the presence of TAM in breast cancer is correlated with poor outcomes104 (figure 2). As the most abundant leukocyte population in mammary tumors,105 breast TAM directly contribute to tumorigenesis by inducing a breast cancer stem cell state, facilitating tumor invasion and metastasis, and promoting angiogenesis.106 Breast TAM can also locally suppress antitumor cytotoxic T cell responses by secreting immune-dampening molecules such as IL-10,15 97 arginase-1,107 and iNOS108 to impair T cell activity. Preventing TAM recruitment with a CSF1R-signaling antagonist had synergistic effects with chemotherapy treatment in a mouse model of breast cancer, resulting in improved survival outcomes.109
Ovarian carcinoma
Ovarian cancer has a high rate of mortality due to its silent onset, rapid metastasis, and advanced stage at the time of diagnosis.110 Metastatic ovarian cancer is characterized by ascites and tumor implants in the peritoneal cavity.111 Unfortunately, results of several single-agent CBT clinical trials in ovarian cancer are discouraging, with objective response rates of 6%–15%.7 As a result, there are no FDA-approved checkpoint inhibitors for ovarian cancer beyond the use of anti-PD-1 (pembrolizumab) for microsatellite instability-high tumors.7 However, compared with monotherapy CBT, dual blockade treatment of anti-PD-1 (nivolumab) and anti-CTLA-4 (ipilimumab) has shown more promising results in the clinic.112 Understanding the contribution of myeloid cells as immune-stimulatory or immunosuppressive cell types can facilitate the rational design of additional combination treatments.
The presence of mature DC in human ovarian carcinomas correlates with a T cell-inflamed TME and improved survival outcomes.113 In patients with human ovarian cancer, tumor expression of B7-H4 and CXCL17 is associated with increased infiltration of mature APC and a proinflammatory TME.114 However, during tumor development, stimulatory DC function can be impacted by the TME to become dysfunctional or immunosuppressive (figure 2). In a mouse model of ovarian cancer, DC induced T cell proliferation at early time points of tumor growth; however, by late time points, DC failed to stimulate T cell expansion and further suppressed the stimulatory function of adjacent immunocompetent DC via production of arginase-1, decreased expression of MHC-II and CD40, and increased expression of PD-L1.115 In line with these observations, early DC depletion could accelerate tumor progression, while late DC depletion delayed tumor progression.115 Similarly, DC isolated from ovarian tumors and ascites at late time points were immunosuppressive and expressed high levels of the inhibitory molecules PD-L1 and PD-1.116 While PD-L1 on DC dampened T cell activation, activation of the PD-1 receptor on DC caused them to produce immune regulatory cytokines and prevented the upregulation of costimulatory markers CD80 and CD86.116
The studies discussed thus far focused on pan-CD11c+ cells and do not delineate the contributions of specific DC subsets to antitumor immunity. While more work is needed to define the roles of cDC1 versus cDC2 in ovarian cancer, the contributions of pDC are better understood. In patients with human ovarian cancer, pDC are abundant117 and exhibit a tolerogenic phenotype (figure 2). pDC in ascites were found to induce tumor angiogenesis via production of TNF-α and IL-8,118 and they could trigger the activation and expansion of ICOS+ FoxP3+ Treg cells.119 Beyond pDC, in primary ovarian tumors, myeloid DC recruit immunosuppressive Treg cells into the tumor by producing CCL22.120 Additionally, a population of inflammatory DC with a suppressive phenotype driven by Satb1 was described in the TME of different models of ovarian cancer.121
The ovarian TME can also actively suppress antitumor immune responses. The constitutive activation of fatty acid synthase in ovarian cancer cells led to lipid accumulation in tumor-infiltrating DC, which impaired their ability to present antigens and prime T cells.122 Ovarian cancer cells can also release extracellular vesicles containing arginase-1 that are internalized by lymphoid DC, resulting in impaired T cell activation.123 While the expression of the TH1-type chemokines, CXCL9 and CXCL10, has been identified to be a strong predictor of improved survival of patients with ovarian cancer,124 ovarian cancer cells can epigenetically silence their own production of CXCL9 and CXCL10 to actively dampen T cell infiltration.125 Furthermore, ovarian tumor-derived VEGF-A, IL-10, and PGE-2 could also induce FasL expression on endothelial cells to selectively kill effector CD8+ T cells.126 Given that cDC1 are a good source of CXCL9 and CXCL10 and can stimulate T cells, strategies to increase their numbers or enhance their function could potentially boost antitumor immune responses against ovarian cancer. Indeed, the antitumor efficacy of Poly (ADP-ribose) polymerase (PARP) inhibition in Brca-1-deficient ovarian cancer was found to be dependent on STING activation of APC, including DC.127 Further studies are needed to clarify the roles of distinct DC subsets in ovarian cancer progression.
The tolerogenic nature of peritoneal macrophages (PM) also promotes aggressive spread of ovarian cancer (figure 2). PM comprise over 30% of all cells in the peritoneal cavity.128 They are highly plastic and depending on the cytokines and growth factors present in the TME, they can be skewed toward an inflammatory M1-like state or an immunosuppressive M2-like state.111 Studies have shown that high amounts of CSF1 secreted by ovarian cancer cells can cause PM to differentiate into CD206+ M2 TAM.129 The resulting increase in the M2/M1 phenotype ratio of TAM has been shown to correlate with decreased survival in patients with ovarian cancer.129 Furthermore, PM can assist cancer cell metastasis by contributing to the formation of tumor-derived multicellular aggregates in the peritoneal fluid.130 PM depletion can reduce both tumor burden and metastasis in a model of ovarian cancer.131 Studies are also ongoing to develop therapeutics to re-polarize M2 TAM into M1 TAM to promote a more proinflammatory TME.111
Pancreatic ductal adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is the most prevalent form of pancreatic cancer and is the fourth leading cause of cancer-related deaths in the world.132 133 Radiation and chemotherapy are minimally effective against PDAC, and surgical resection is the only curative treatment.132 However, as most cases of pancreatic cancer are detected only at the locally advanced or metastatic stages, complete surgical resection is often impossible. Furthermore in the clinic, PDAC has been shown to be non-responsive to monotherapy CBT, although efforts are underway to evaluate responses with combination immunotherapies.133 Due to late detection and limited treatment options, the 5-year survival rate of pancreatic cancer is dismal, less than 5%.132 133
CD103+ cDC1 and CD11b+ DC comprise the two main subsets of tissue-resident DC in the pancreatic islets in both steady state and inflammatory contexts.134 Islet CD103+ DC are migratory and can prime T cells in the draining lymph nodes with β cell-derived antigens.134 135 In contrast, while CD11b+ DC/moDC can efficiently take up antigen, they have a low capacity to activate T cells, and therefore, appear more related to macrophages by function.134 CD11b+ DC/moDC greatly outnumber CD103+ DC at steady state, and the balance of activities of these two subsets determines whether tolerance or inflammation is induced.134
However, DC are poorly infiltrated in human PDAC and, if present, have been reported to be spatially restricted to the tumor margin.136 Consistent with this observation, cDC were also reported to be rare in the KPC murine model of PDAC.71 The few tumor-infiltrating cDC1 expressed low levels of costimulatory markers and were weakly stimulatory in ex vivo coculture assays with T cells.71 The scarcity of cDC1 in PDAC is likely a consequence of an active immune evasion mechanism by the tumor. PDAC-secreted granulocyte colony-stimulating factor caused downregulation of the transcription factor IRF8 that is necessary for cDC1 development.137 The resultant decrease in numbers of cDC1 progenitors and cDC1s in the bone marrow and peripheral blood impaired antitumor CD8+ T cell responses. DC that do infiltrate (typically CD11b+) PDAC tumors are often immature and functionally impaired or are immunosuppressive in nature. In an orthotopic mouse model of PDAC, IL-23-producing and TGF-β-producing CD11b+ DC comprised the greatest fraction of tumor-infiltrating DC.138 This DC population induced the differentiation of tolerogenic FoxP3neg CD4+ T cells that produced IL-10, IL-17, and IFNγ and promoted tumor outgrowth. Depleting these immunosuppressive DC resulted in decreased tumor burden. FoxP3+ Treg cells can also engage with DC in the PDAC TME, resulting in suppression of DC function via downregulation of MHC-II expression and costimulatory molecules CD40 and CD86 over time.139 The combination of Flt3L treatment with a CD40 or STING agonist could overcome the deficiency of mature, functional cDC in PDAC tumors and promoted a TH1 microenvironment that resulted in antitumor immunity.71
While stimulatory DC are scarce in PDAC, TAM are highly abundant, comprising one of the dominant immunosuppressive myeloid populations in PDAC tumors140 (figure 2). PDAC TAM generally correlate with poor prognosis and perform immune-inhibitory functions.141 Embryonic-derived resident TAM in PDAC secrete profibrotic factors and have reduced surface expression of MHC-II molecules, reflecting an impaired ability to stimulate T cell responses.141 Given their protumorigenic association, the inhibition or targeted depletion of TAM in PDAC has proven to be effective in controlling tumor burden and reducing the development of high-grade invasive tumors, largely by reprogramming the PDAC tumor stroma to be more proinflammatory and enhancing adaptive immune responses.141 142
Liver metastasis
Liver is one of the most permissive organs for metastasis, and as such, the majority of liver lesions are secondary metastatic tumors.143 (Primary hepatocellular carcinoma will not be discussed here due to its viral etiology: hepatitis B and C virus infections are responsible for over 75% of cases143.) Liver metastases respond poorly to CBT, and their presence correlates with reduced therapy effectiveness in patients with melanoma and NSCLC.5 CBT resistance of liver metastases is not surprising given the tolerogenic microenvironment of the liver, needed to avoid excessive tissue injury from continuous exposure to nutrients and microbial antigens from the gut. Tolerance is established by liver-resident cells via many mechanisms, including production of IL-10 and TGF-β, induction of Treg cells, inhibition of CD8+ T cell priming, and direct engulfment and degradation of autoreactive T cells.144 145 Although liver APC comprise both conventional and unconventional cell types, we will only discuss DC and macrophages here (for a comprehensive review of liver APC refer to study by Horst et al 144 and Knolle145).
Due to the tolerogenic microenvironment, at baseline, liver DC are predominantly functionally immature. Compared with immature blood DC, liver DC secrete high levels of IL-10 and low levels of IL-12, are poor antigen presenters and weak stimulators of CD4+ T cells, can polarize TH2 cell differentiation, and induce Treg cells.146 Liver stromal cells promote differentiation of DC progenitors into a tolerogenic DC subset, which is capable of secreting IL-10 and PGE-2, inhibiting activation of naive T cells and inducing apoptosis of activated T cells.147 Characterization of steady state DC reveals the presence of CD103+ cDC1, CD11b+ cDC2, and pDC subsets in both mouse and human livers.148–150 At baseline, liver cDC1 express lower levels of PD-L1 compared with cDC2, suggesting that they might be less inhibitory.148 Studies of pDC suggest that this subset is more tolerogenic in the liver than in the spleen, as liver pDC express higher levels of PD-L1, lower levels of CD80 and CD86, and produce more IL-10 and less IL-12p70.151 Interestingly, stratifying liver DC based on lipid content suggests that low-lipid DC are tolerogenic and induce Treg cells, while high-lipid DC are immune-stimulatory and can activate CD8+ T cells, Natural Killer (NK) cells, and Natural Killer T (NKT) cells, implying that DC maturation in the liver can be metabolically regulated.152
Liver metastases become enriched in immunosuppressive DC. In a preclinical model of metastatic pancreatic cancer, CD11b+ moDC were found to accumulate at liver metastases and promote tumorigenesis by secreting IL-6, TNF-α, and CCL2, upregulating PD-L1, PD-L2, and ICOS-L and thus supporting Treg cell expansion.153 Both selective depletion of these inhibitory DC and PD-L2 blockade can reverse Treg-mediated metastatic progression by inducing CD8+ T cell responses.153 In patients with liver metastases from colorectal cancer, pDC infiltrate tumors, upregulate ICOS-L, and thereby induce accumulation of IL-10-producing CD4+FoxP3neg suppressive T cells.154 Further research is needed to elucidate the specific contribution of each hepatic DC subset to CBT resistance of liver metastases.
Kupffer cells (KC) are the most abundant subtype among tissue-resident macrophages both in the liver and in the whole body and are largely immunosuppressive.144 145 While KC on their own are capable of effectively priming naive CD8+ T cells locally in the liver, the presence of viral antigen-presenting hepatocytes tolerizes their stimulatory capacity.155 The resulting CD8+ T cells proliferate but exhibit dysfunction, as they produce low levels of IFNγ and display poor effector functions.155 Moreover, KC can promote the formation of a premetastatic niche for pancreatic cancer, by taking up tumor-derived exosomes and inducing TGF-β and increased fibronectin production in hepatic stellate cells, thereby priming the liver for metastasis.156 In a study focused on colorectal liver metastases, CCR5 blockade induced TAM repolarization characterized by reduced matrix metalloproteinase secretion, increased IFNα production, and led to protective antitumor effects in patients.157
Conclusion
Tissue-specific DC and macrophages can dominantly impact antitumor immunity and influence responses to CBT and other therapies. While it is impossible to make absolute statements, we can define overarching patterns observed from surveying the tissue-specific myeloid composition of CBT-sensitive and CBT-refractive carcinomas (figure 2; tables 1–2).
The major DC subsets, namely cDC1, cDC2, and pDC, vary in their degree of infiltration in different tumors. Although the functions of these DC subsets are largely context-dependent, the role of cDC1 across carcinomas is most clearly defined: they are the primary inducers of protective antigen-specific CD8+ T cell responses. The functions of cDC2/moDC and pDC can also be stimulatory, but in the TME, they are often skewed toward tolerance, as observed in lung, breast, and pancreatic cancer models. Tumors that are highly infiltrated with stimulatory DC (such as NSCLC) are generally associated with a T cell-inflamed TME and improved treatment outcomes when compared with those that are poorly infiltrated (such as PDAC). However, exclusion or functional suppression of stimulatory DC is a common mechanism of immune evasion by the tumor. Consequently, treatment strategies to boost DC numbers and stimulatory capacity can result in decreased tumor burden even in an immune desert like PDAC. While the phenotypes of DC subsets are largely conserved between different tumors types, certain functional DC characteristics are tissue-specific, such as the substantial level of baseline tolerance of liver DC. This observation warrants a deeper investigation into the tissue-specific differences in DC function, in order to allow for the successful development of restimulation strategies for DC residing at distinct tissue sites.
In contrast to rare DC, TAM are abundant in all tumor types examined. They exhibit tissue-specific plasticity, and their stimulatory (M1-like) or suppressive (M2-like) function depends on environmental signals. In all six carcinomas, however, TAM appear to be immunosuppressive, protumorigenic, and associated with poor outcomes. TAM depletion, functional inhibition, or repolarization led to improved outcomes in several preclinical models spanning multiple cancer types and has translational implications.
In summary, modulation of the myeloid APC compartment has the potential to boost antitumor immunity. Studies of tissue-specific DC and macrophages will uncover novel ways to remodel the myeloid compartment and provide new avenues to overcome the limitations of currently available treatments.
Acknowledgments
The authors thank the NIH Pre-Doctoral Training Grant T32GM007287 and the Pew Charitable Trust and Howard S (1953) and Linda B Stern for funding.
References
Footnotes
MZ and ED contributed equally.
Contributors MZ, ED, and SS conceived the outline; MZ and ED wrote the manuscript with supervision from SS.
Funding MZ and ED are supported by the NIH Pre-Doctoral Training Grant T32GM007287 and SS is a Pew-Steward Scholar of the Pew Charitable Trust and holds the Howard S (1953) and Linda B Stern Career Development Professorship.
Competing interests SS is a consultant or SAB member of Arcus Biosciences, Dragonfly Therapeutics, TAKEDA, Merck, Ribon Therapeutics, Replimune, and Tango Therapeutics, but these activities are not in conflict with the presented data.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.