Article Text

Abstract
Background The cancer-testis antigen MAGE-A4 is an attractive target for T-cell-based immunotherapy, especially for indications with unmet clinical need like non-small cell lung or triple-negative breast cancer.
Methods An unbiased CD137-based sorting approach was first used to identify an immunogenic MAGE-A4-derived epitope (GVYDGREHTV) that was properly processed and presented on human leukocyte antigen (HLA)-A2 molecules encoded by the HLA-A*02:01 allele. To isolate high-avidity T cells via subsequent multimer sorting, an in vitro priming approach using HLA-A2-negative donors was conducted to bypass central tolerance to this self-antigen. Pre-clinical parameters of safety and activity were assessed in a comprehensive set of in vitro and in vivo studies.
Results A MAGE-A4-reactive, HLA-A2-restricted T-cell receptor (TCR) was isolated from primed T cells of an HLA-A2-negative donor. The respective TCR-T-cell (TCR-T) product bbT485 was demonstrated pre-clinically to have a favorable safety profile and superior in vivo potency compared with TCR-Ts expressing a TCR derived from a tolerized T-cell repertoire to self-antigens. This natural high-avidity TCR was found to be CD8 co-receptor independent, allowing effector functions to be elicited in transgenic CD4+ T helper cells. These CD4+ TCR-Ts supported an anti-tumor response by direct killing of MAGE-A4-positive tumor cells and upregulated hallmarks associated with helper function, such as CD154 expression and release of key cytokines on tumor-specific stimulation.
Conclusion The extensive pre-clinical assessment of safety and in vivo potency of bbT485 provide the basis for its use in TCR-T immunotherapy studies. The ability of this non-mutated high-avidity, co-receptor-independent TCR to activate CD8+ and CD4+ T cells could potentially provide enhanced cellular responses in the clinical setting through the induction of functionally diverse T-cell subsets that goes beyond what is currently tested in the clinic.
- immunotherapy
- adoptive
- antigen presentation
- CD8-positive T-lymphocytes
- CD4-positive T-lymphocytes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Empowering a patient’s own immune system to recognize and fight cancer has been one of the most challenging but also one of the most promising goals of anti-cancer immunotherapy. Patients with cancer as well as healthy individuals may have naturally occurring T cells that are capable of distinguishing healthy from cancerous cells based on recognition of tumor-associated antigens (TAAs).1 Unfortunately, not all patients have T cells that effectively recognize their tumors and, even when tumor-specific T cells exist, they are often poorly activated and not expanded to the numbers needed to facilitate tumor eradication.2 Engineered autologous TCR-T immunotherapy involves genetically modifying and equipping large numbers of patient-derived T cells with a selected TCR recognizing a defined TAA-derived epitope that is presented by the patient’s tumor cells.3 Compared with chimeric antigen receptors (CARs) which only recognize extracellular epitopes, TCRs have the advantage of additionally recognizing a larger pool of epitopes derived from intracellular proteins presented on HLA molecules.4 Successful adoptive transfer of TCR-Ts has been shown for several TAAs.5 6
However, choice of the right target antigen is a critical step for success of TCR-T immunotherapies.7 Expression of the cancer-testis antigen melanoma-associated antigen (MAGE)-A4 in adult healthy tissues is limited to immune-privileged sites.8 9 Furthermore, MAGE-A4 is highly expressed in a variety of cancer indications with unmet clinical need, such as non-small cell lung cancer (NSCLC), head and neck, ovarian, triple-negative breast, and gastroesophageal cancers, thereby representing a suitable target antigen for TCR-T immunotherapy.10 11
To overcome tumor burden, TCR-Ts must have high avidity toward the peptide/HLA complex (pHLA) present on tumor cells.12 However, high-avidity T cells recognizing TAAs presented on self-HLAs are rarely present in the human T-cell repertoire, as these cells are largely eliminated during the process of negative selection in the thymus.13 14 In vitro affinity maturation of autologous-derived TCRs can correct for low affinity but often results in decreased peptide specificity, increasing the risk for off-target toxicities.15 16
The process of central tolerance is bypassed when TCRs are isolated from certain humanized mouse models17 or when they are derived from donor T cells primed using mismatched HLA molecules (ie, allo-restricted peptide presentation)18 since negative selection is limited to self-HLA molecules.13 19
CD8 molecules are known to significantly increase the TCR-based recognition of peptides presented by HLA class I molecules.20 This CD8 dependency, however, can be overcome by certain higher affinity TCRs if their binding to the pHLA complex is greater than the binding affinity of HLA class I to CD8.21 22 With such TCRs, desired functions of TCRs can also be transferred to CD4+ T cells to facilitate direct tumor cell killing,23 antigen-specific help during several phases of an immune response, and promotion of long-term in vivo persistence of tumor-reactive CD8+ T cells.24 25
Herein we describe the potency and specificity of a CD8 co-receptor independent TCR targeting MAGE-A4 that was generated from an in vitro priming approach that leverages HLA-mismatched donors to avoid immunologic tolerance.
Materials and methods
Immunohistochemistry
Human tumor biopsies (n=481) of different histologic subtype were obtained as formalin-fixed paraffin-embedded blocks. MAGE-A4-negative A549 cells and MAGE-A4-transduced A549 cells were used as controls, and their MAGE-A4 expression status was confirmed via WESTM, qPCR, and/or Nanostring experiments (data not shown). All blocks including the cell pellets and tissues were processed to slides (5 µm thick sections) and stained with a mouse anti-human MAGE-A4 monoclonal antibody (ThermoFisher, OTI1F9, cat# MA5-26118, 0.04 µg/mL) or an irrelevant isotype control (mouse IgG2a, Abcam ab#18413) according to standard protocols using a Biocare Intellipath autostaining system. An exception was triple-negative breast cancer, which was done manually. Slides were developed with DAB and counterstained with hematoxylin.
Slides were evaluated by the study pathologist using a standard bright-field microscope. The intensity of tumor cell immunoreactivity was scored 0=no, 1=low/minimal, 2=moderate, and 3=marked membranous and/or cytoplasmic immunoreactivity. The percentages of cells assigned to each score were estimated and an H-score assigned: H-score=[1*(%cells score1)+2*(%cells score2)+3*(%cells score3)]. Images were captured using the Panoramic 3D Histech scanner and CaseViewer digital pathology software (3DHISTECH).
Cells
K562, T2 (DSMZ), LCLs (fredhutch.org), MelA375, NCI-H1755, UACC62, NCI-H1703, U266, A549, and NCI-H520 (ATCC) were maintained in RPMI-1640 (+10% FCS, 1% NEAA, 1% L-glutamine, 1% sodium-pyruvate). 647V and HEK293FT were maintained in DMEM (+10% FCS, 1% NEAA, 1% L-glutamine, 1% sodium pyruvate). MCF7 were maintained in DMEM (+10% FCS, 0.01 mg/mL insulin). SAOS2 were maintained in McCoy’s 5A (+15% FCS). NCI-H2023 were maintained in DMEM/F-12 (+5% FCS, 5 µg/mL insulin, 10 µg/mL transferrin, 30 nM sodium selenite, 10 nM hydrocortisone, 2 mM L-glutamine, 10 nM beta-estradiol). All cell lines were tested for mycoplasma negativity by PCR (Venor GeM-Classic; Minerva Biolabs).
iCell cardiomyocytes2, endothelial cells, hepatocytes 2.0, astrocytes, and GABAneurons were purchased from Cellular Dynamics. Normal human lung fibroblasts and bronchial epithelial cells were purchased from Lonza and renal cortical epithelial cells from PromoCell. All cell types were cultured according to manufacturers’ instructions.
Retroviral transduction of cells was done using plasmids encoding HLA-A*02:01, CD86, and either GFP alone or fused to one of the codon optimized target antigens via a porcine teschovirus-1 2A (P2A) sequence. Transduced cells were enriched by FACS for GFP expression.
Flow cytometry and cell sorting
Staining of cell surface proteins was done according to standard protocols.19 26 Intracellular staining to identify Granzyme B was done using the FoxP3/transcription buffer set (ThermoFisher, 00-5523-00) according to the manufacturer’s protocol. The following fluorochrome-labeled antibodies were used: anti-human CD8 (RPA-T8, Becton Dickinson, 560347), CD4 (RPA-T8, Becton Dickinson, 7137857), Granzyme B (GB11, BioLegend, 515405), HLA-A2 (BB7.2, Becton Dickinson, 561341), CD137 (4B4-1, Becton Dickinson, 550890), CD154 (TRAP1, Becton Dickinson, 555700).
CD8+ T cells specific for MAGE-A4 in context of HLA-A2 were identified by multimer staining. Fluorochrome-labeled pentamers were custom-synthesized (ProImmune) and staining was done according to the manufacturer’s instructions, also including staining for CD8 and CD4 when appropriate. Cell sorting was conducted using a FACSAria Fusion flow cytometer (Becton Dickinson) or a SH800S cell sorter (Sony).
Production and electroporation of in vitro transcribed RNA (ivt-RNA)
HLA-A*02:01, full-length MAGE-A4 or midi-gene constructs containing up to 200 base pairs (bp) of the original gene sequence 5′ and 3′ of the respective peptide-coding region were custom synthesized as codon-optimized DNA at GeneArt (ThermoFisher). Fragments were cloned into the pGEM vector using the seamless cloning technique.27 ivt-RNA was produced and purified using the mMESSAGE mMACHINE T7 Kit (Life Technologies, AM1344) and RNeasy Mini Kit (Qiagen, 74106) following manufacturers’ instructions. Electroporation was done using a Gene Pulser XCell (BioRad) with an exponential protocol (300 V, 150 µF).
Isolation of MAGE-A4-reactive T-cell clones
T cells were stimulated as previously described.19 26 In brief, blood was drawn from healthy donors after obtaining informed consent in accordance with company and governmental guidelines and approved by the ethics commission of the Bavarian State Chamber of Medicine. Monocyte-derived mature dendritic cells (mDC) were generated as previously described.28 mDC were harvested on day 8 and transfected with 20 µg ivt-RNA encoding for MAGE-A4 in HLA-A*02:01-positive donors, or 20 µg ivt-RNA encoding for MAGE-A4 together with 20 µg ivt-RNA encoding for HLA-A*02:01 for HLA-A2-negative donors. CD8+ T cells were enriched from PBMC by negative selection (CD8+ T cell Isolation Kit, Miltenyi Biotec, 130-094-156) and co-cultured with the transfected mDC (10:1) in T-cell medium (TCM) consisting of RPMI-1640 (+10% heat-inactivated human serum, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 50 µM β-mercaptoethanol, 100 U/mL penicillin/streptomycin). Cell cultures were supplemented with 5 ng/mL IL-7 (PeproTech, AF-200–07) at day 0 and propagated by addition of 50 IU/mL IL-2 (Novartis, DA0286AAF) every 2–3 days starting at day 1.
CD137-based enrichment was conducted for HLA-A*02:01-positive donors as follows: T cells were propagated 12 days before co-culture with K562 cells stably expressing HLA-A2, CD86 (A2+K562), and an irrelevant protein (10:1) in TCM (+20 IU/mL IL-2). After 16 hours, cells were stained for CD137 and the CD137-negative fraction was sorted and cultured in TCM. CD137-negative cells were co-cultured in TCM with irradiated (100 Gy) A2+K562 cells expressing MAGE-A4 (10:1). After 16 hours, cells were stained for CD137 and single cell sorting of the CD137-positive fraction was conducted. Similarly, single cell-sorting using multimers was performed 8 days after T-cell propagation using material of HLA-A*02:01-negative donors as outlined previously. Single cell sorting was done in 96 wells containing 200 µL re-stimulation cocktail (0.5–4×105 LAZ-388 cells/mL irradiated at 150 Gy and 0.5–1×106 PBMC/mL irradiated at 50 Gy in TCM supplemented with 50 IU/mL IL-2 and 32 ng/mL anti-CD3 antibody (OKT-3, Helmholtz Center Munich, custom synthesis)).
Ten days after re-stimulation, T-cell clones were co-cultured overnight with A2+K562 cells expressing either MAGE-A4 or an irrelevant protein. Release of IFN-γ was assessed as described later and T-cell clones displaying a MAGE-A4-specific reaction pattern were further propagated in 2 mL TCM containing 5×105 PBMC/mL irradiated at 50 Gy, 5×105 A2+K562 cells transduced with MAGE-A4/mL irradiated at 100 Gy, and supplemented with 100 IU/mL IL-2 and 250 ng/mL phytohaemagglutinin (Remel, R30852801). Medium was fully replaced after 5 days by TCM and cultures were propagated by addition of 50 IU/mL IL-2 every 2–3 days.
T-cell receptor sequencing
For identification of TCR-α and TCR-β chains, next-generation sequencing was conducted with established standard protocols for analysis with the MiSeq system (Illumina). The manufacturer’s recommendations were followed using the Dynabeads mRNA DIRECT Kit (Life Technologies, 61012), SMART Scribe reverse transcriptase (Takara, 639538), and the MiSeq V3 Kit (Illumina, MS-102-3003).
Cloning, virus production, and transduction of T cells
Wildtype TCR-β and TCR-α chain sequences were linked by P2A, codon-optimized, and cloned in the replication defective, self-inactivating third-generation HIV-1-based lentiviral vector that harbors the murine leukemia virus–derived MND promoter to drive expression of the TCR.29 VSV-G pseudotyped lentiviral particles were produced by transient transfection of HEK293FT cells with the vector plasmid and packaging plasmids encoding GAG/POL, REV, and VSV-G, purified via chromatography and formulated before storage at ≤−65°C. PBMCs were isolated from leukapheresis material (Key Biologics) of healthy donors and cryopreserved. Thawed PBMCs were activated with soluble OKT3 (Miltenyi Biotec), and human anti-CD28 (Miltenyi Biotec, clone 15E8), in T-cell growth medium (TCGM) consisting of X-VIVO 15 medium (Lonza) supplemented with 5% human serum type AB (Valley Biomedical), 2 mM of GlutaMAX-I (Gibco), 10 mM HEPES buffer solution (Gibco), and 250 IU/mL of IL-2 (CellGenix GmbH). The next day, T cells were transduced and expanded for 10 days before bulk sorting and re-stimulation of multimer-positive cells was done as described previously. Bulk sorting of TCR-transduced PBL was done by sorting up to 2×105 cells and subsequent propagation in 5–15 mL re-stimulation cocktail. Medium of bulk-sorted re-stimulation cultures was fully replaced after 72 hours by TCM and cultures were propagated by addition of 50 IU/mL IL-2 every 2–3 days for a period of 10 days before use in co-culture assays.
For murinized TCR sequences, the constant regions of both TCR chains were exchanged by their murine counterparts to increase the stability of the TCR.30 Codon-optimized sequences were cloned in the γ-retroviral vector plasmid pES12.6. Virus production and transduction of T cells was done as described.19
Co-culture assays
Target cells were seeded into 96-well U-bottom plates (1–2×104 cells/well). Effector cells (1–2×104 cells/well) were added and the co-culture plates (E:T ratio of 1:1) were incubated for 16–24 hours or 6 hours to determine CD154 expression. Duplicates or quadruplicates were tested for all conditions.
Target cells were individually loaded with peptides (Pepd4LS) at a final concentration of 10−5 M. After 1.5-hour incubation at 37°C, peptide-loaded target cells were washed with PBS and resuspended in RPMI-1640 (+10% FCS, 1% NEAA, 2 mM L-glutamine, 1 mM sodium pyruvate) for co-culture. For analysis of functional avidity, T2 cells were loaded with titrated amounts of peptide. EC50 values were determined with a variable slope model Y=100/(1+10∧((LogEC50−X)*HillSlope)).
Cytokine release assays
IFN-γ and IL-2 release was assessed in co-culture supernatants and cytokine concentrations were determined using ELISA kits (ThermoFisher, 88-7316-77; Becton Dickinson, 555 142; or Becton Dickinson, 555190). The OD measurement was performed using a Multiskan Microplate-Photometer (ThermoFisher). Background-corrected OD values were used for extrapolation using a third-degree polynomial.
Polyfunctional cytokine profiles (IL-2, IL-4, IL-5, IL-21, GM-CSF, IFN-γ, and TNF-α) in co-culture supernatants were determined with a customized kit (Luminex ProcartaPlex platform; ThermoFisher) according to the manufacturer’s instructions.
Cytotoxicity assay
Cell lysis was assessed with an IncuCyte Zoom 2CLR (Sartorius) following the manufacturer’s protocols for real-time quantitative live cell imaging. Tumor cells were transduced with NucLightRed (Sartorius) and seeded into 96-well flat-bottom plates 24 hours before addition of T cells. Plates were scanned at regular intervals and the number of NucLightRed-labeled cells was determined with the IncuCyte Software (V.2018A; Sartorius).
Xenograft models
Female NOD-Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG; Jackson Laboratories) mice received subcutaneous injections of 5×106 MAGE-A4/HLA-A*02:01-positive MelA375 melanoma tumor cells. Eight days post tumor implantation, mice were randomized into four groups of five mice. Next day, groups received a single intravenous injection of vehicle or 3.5×107 transgenic TCR-positive T cells or an equivalent number of untransduced T cells. Mice were monitored until day 37 post T-cell injection and tumor volume was measured via calipers twice a week by calculating length×width×height×π/6. All procedures and housing were compliant with the Guide for the Care and Use of Laboratory Animals, eighth edition and were approved by bluebird bio’s IACUC.
Results
Tumor-associated antigen MAGE-A4 harbors an immunogenic candidate T-cell epitope
Multiple human tumor samples of different histologic subtype were used for immunohistochemistry staining using an anti-MAGE-A4 antibody (online supplemental figure 1). Around 18% of triple-negative breast carcinoma (TNBC), 16% of head and neck squamous cell carcinoma (HNSCC), 18% of non-small cell lung carcinoma (NSCLC), and 15% of ovarian cancer samples received an H-score equal to or above 100, indicating robust MAGE-A4 expression (figure 1A). The prominent expression of MAGE-A4 in several solid tumor indications makes it a promising target for adoptive cell therapy with TCR-Ts.
Supplemental material
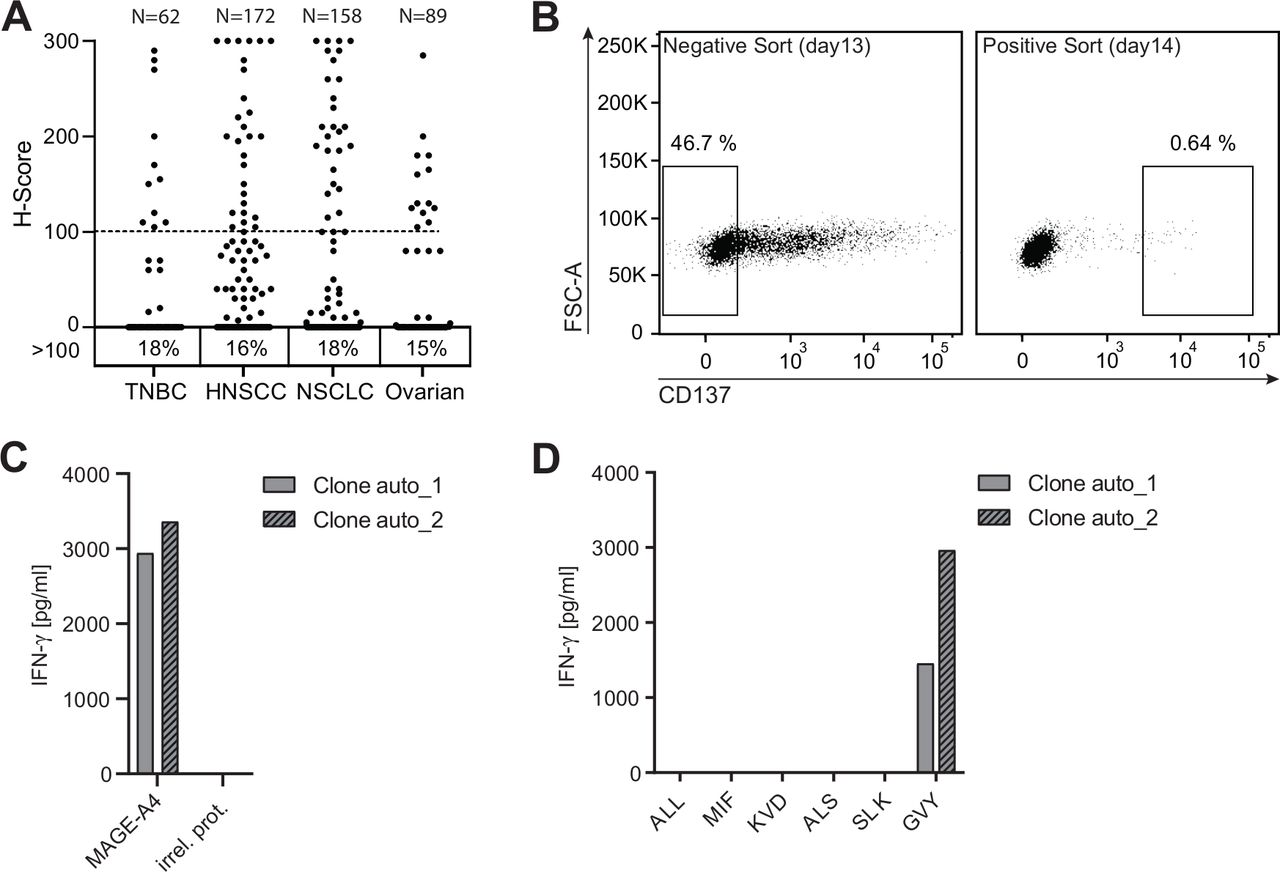
Tumor-associated antigen MAGE-A4 harbors an immunogenic candidate T-cell epitope. (A) Formalin-fixed paraffin-embedded human tumors of different histologic subtype (TNBC n=62, HNSCC n=172, NSCLC n=158, ovarian n=89) were stained with an anti-human MAGE-A4 antibody and evaluated by a study pathologist. The intensity of staining was scored based on a 3-point scale, the percentages of cells assigned to each score were estimated, and an H-score was assigned. Numbers below graph indicate the percentage of samples with an H-score ≥100. (B) CD8+ T cells from an HLA-A*02:01-positive healthy donor were expanded for 12 days by stimulation with autologous monocyte-derived dendritic cells electroporated with ivt-RNA encoding for MAGE-A4. Left dot plot depicts the gating strategy of expanded CD8+ T cells 16 hours after co-culture with MAGE-A4-negative A2+K562 cells for CD137-negative bulk sorting. CD137-negative sorted cells were co-cultured with A2+K562 cells transduced with MAGE-A4; 16 hours later, CD137-positive cells (right) were single cell sorted. (C,D) Single cell sort–derived T-cell clones were expanded for 15 days and tested for reactivity toward MAGE-A4. Shown are bar graphs displaying IFN-γ concentrations in supernatants of two positive T-cell clones 16 hours after co-culture with target cells for screening. (C) Co-culture with A2+K562 cells transduced with MAGE-A4 or an irrelevant protein (irrel. prot.). (D) Co-culture with A2+K562 cells pulsed with different candidate peptides.
The identification of MAGE-A4-derived immunogenic peptides presented on HLA-A*02:01-encoded molecules was achieved by stimulating CD8+ T cells from four HLA-A*02:01-positive healthy donors with autologous DCs overexpressing MAGE-A4. MAGE-A4-reactive T cells were enriched using a sorting approach based on the activation-induced marker CD137 (alias 4-1BB)31 allowing the identification of reactive T-cell clones bypassing the requirement for defined pHLA-multimers.32 To increase the specificity of CD137-based enrichment, CD8+ T cells were first co-cultured overnight with A2+K562 cells transduced with an irrelevant control protein. Since A2+K562 cells are endogenously negative for MAGE-A4, non-specifically activated CD8+ T cells were depleted by conducting a bulk-sort for CD137-negative T cells (figure 1B; left plot). Thereafter, enriched CD137-negative T cells were co-cultured with A2+K562 cells transduced with MAGE-A4 overnight, and CD137-positive T cells were sorted as single cells (figure 1B; right plot) and expanded.
MAGE-A4-reactive T-cell clones were identified by recognition of MAGE-A4-positive A2+K562 cells without response to the negative control cells (figure 1C). To identify recognized epitopes, individual T-cell clones were tested for response to a panel of predicted or previously reported MAGE-A4-derived peptides with sequences not found in other MAGE family members (table 1). The 10-mer peptide GVYDGREHTV (hereafter GVY)33 was found to be dominantly recognized, as indicated by IFN-γ secretion of two T-cell clones derived from one donor (figure 1D). Interestingly, the predicted affinity of GVY toward HLA-A2 is comparatively low,34 35 highlighting the benefit of epitope identification using a prediction-independent process.
MAGE-A4-derived HLA-A*02:01-restricted candidate epitopes
bbT485 cells expressing an allo-derived TCR exhibit optimal epitope binding characteristics
As high-avidity T cells specific for self-peptides presented on self-HLAs are eliminated by negative selection,13 a previously described allogeneic priming approach was used to isolate high-avidity clones from T-cell repertoires that have not undergone negative selection.19 In this context, CD8+ T cells from six HLA-A2-negative donors were activated with autologous mDCs overexpressing MAGE-A4 and HLA-A*02:01-encoded molecules. Activated CD8+ T cells were used for single-cell sorting with fluorochrome-labeled HLA-A2-multimers loaded with the GVY peptide. Derived T-cell clones were tested for MAGE-A4 reactivity (data not shown).
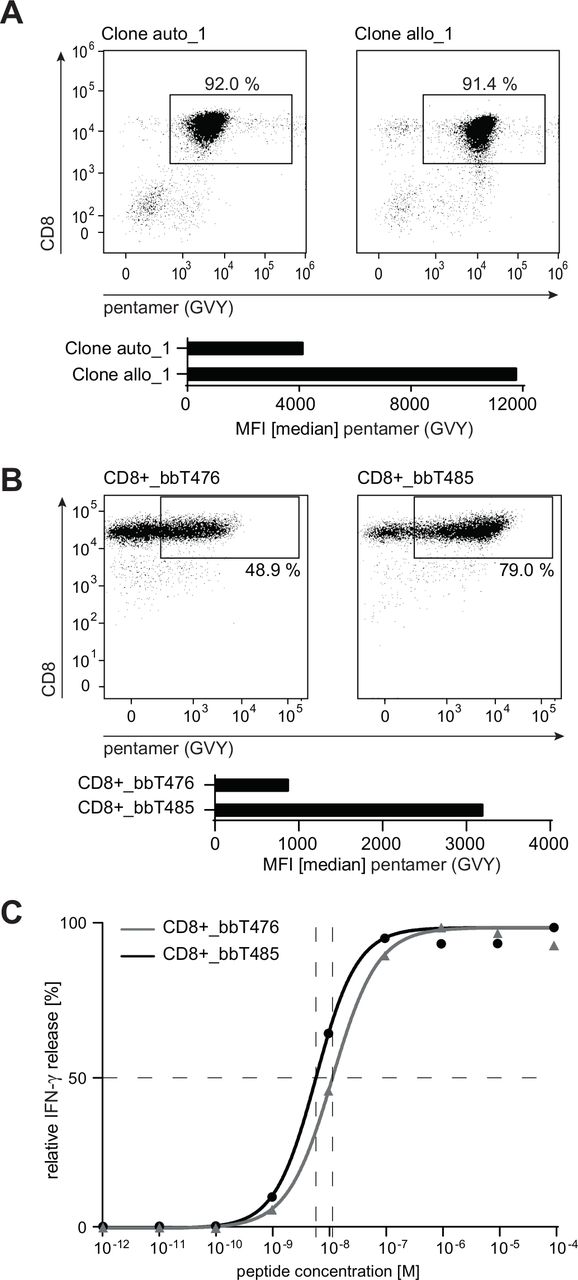
TCR sequences of the most potent allo-derived T-cell clone (clone allo_1) and its autologous-derived counterpart (clone auto_1) isolated by CD137-based sorting were obtained by next-generation sequencing. Rearranged TCR α and β V(D)J chains were cloned using either human or murine constant regions. Use of murine constant regions ensures minimal mispairing of recombinant TCR chains with endogenous TCR α and β chains and thereby increases surface expression of murinized transgenic TCRs.30 All TCR sequence variants were transduced into PBLs of healthy donors and the respective TCR-T products were designated bbT476 for the autologous-derived TCR and bbT485 for the allo-derived TCR. In vitro expanded CD8+ and GVY/HLA-A2-multimer-positive T cells of both products exhibited an effector memory like phenotype (CCR7neg, CD45RAneg/low; data not shown) and comparable proliferation capacities. Essential assays investigating TCR safety and potency were conducted with both constant region sequences and results were comparable between murinized (data not shown) and fully human TCRs.
Flow cytometry evaluating GVY/HLA-multimer binding was performed with the original T-cell clones and TCR-Ts. When compared with T cells expressing the autologous-derived TCR, median fluorescence intensities (MFIs) of multimer-positive cells expressing the allo-derived TCR were higher in both the original T-cell clone (figure 2A) and in T cells transduced with human (figure 2B) and murinized TCRs (online supplemental figure 2).
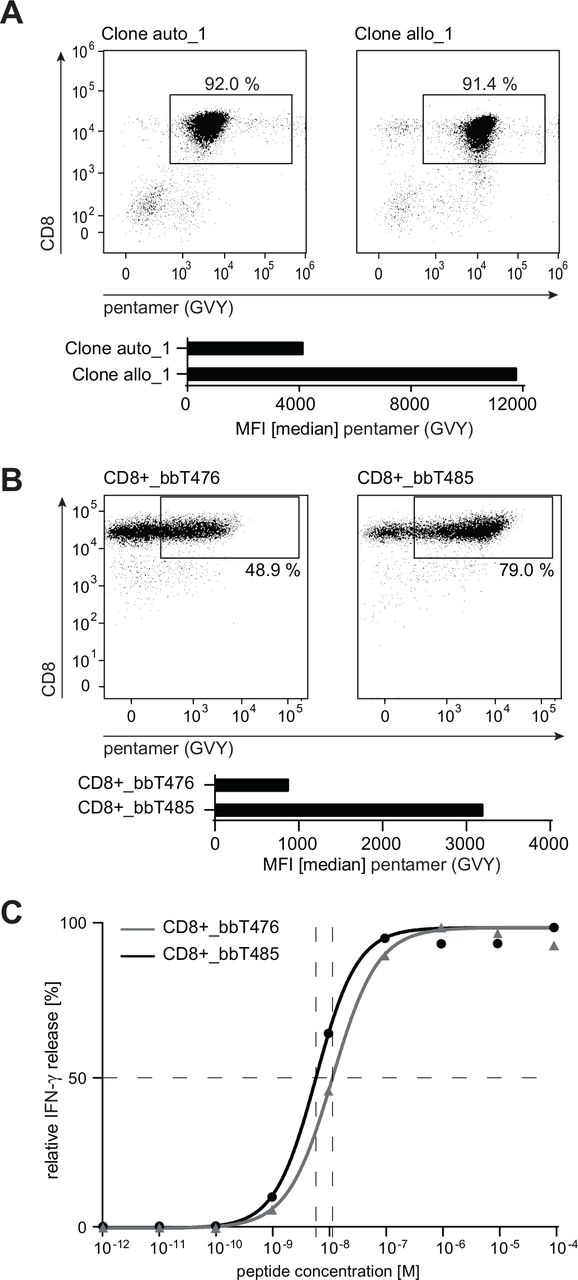
bbT485 TCR-Ts expressing an allo-derived TCR exhibits superior epitope binding characteristics compared with bbT476 expressing the auto-derived TCR. (A,B) Dot plots indicating the percentage of HLA-A2-GVYDGREHTV pentamer-positive CD8+ T cells and bar graphs showing median fluorescence intensity (MFI) within respective gates. (A) Analysis of the original T-cell clones. (B) Analysis of healthy donor-derived PBL transduced and enriched to express either the auto-derived or the allo-derived TCR. (C) Dose–response curves determining the EC50 value for bbT476 or bbT485 CD8+ T cells. Shown are the relative individual values and the non-linear regression curve of IFN-γ determined by ELISA 16 hours after co-culture with T2 cells pulsed with different concentrations of the GVY peptide. (B,C) Data shown are representative of 3 different donors for each tested TCR.
Since differences in measured MFIs do not necessarily reflect the potency of a TCR for tumor rejection,36 the functional avidity of both TCRs was compared. T2 cells loaded with varying concentrations of GVY peptide were used to measure cytokine secretion of the TCR-Ts at 24 hours. The peptide concentration required to induce half-maximal IFN-γ secretion (EC50 values) was lower for bbT485 CD8+ T cells expressing the allo-derived TCR when compared with bbT476 CD8+ T cells expressing the autologous-derived TCR (figure 2C).
bbT476 and bbT485 TCR-Ts both show good in vitro safety profiles
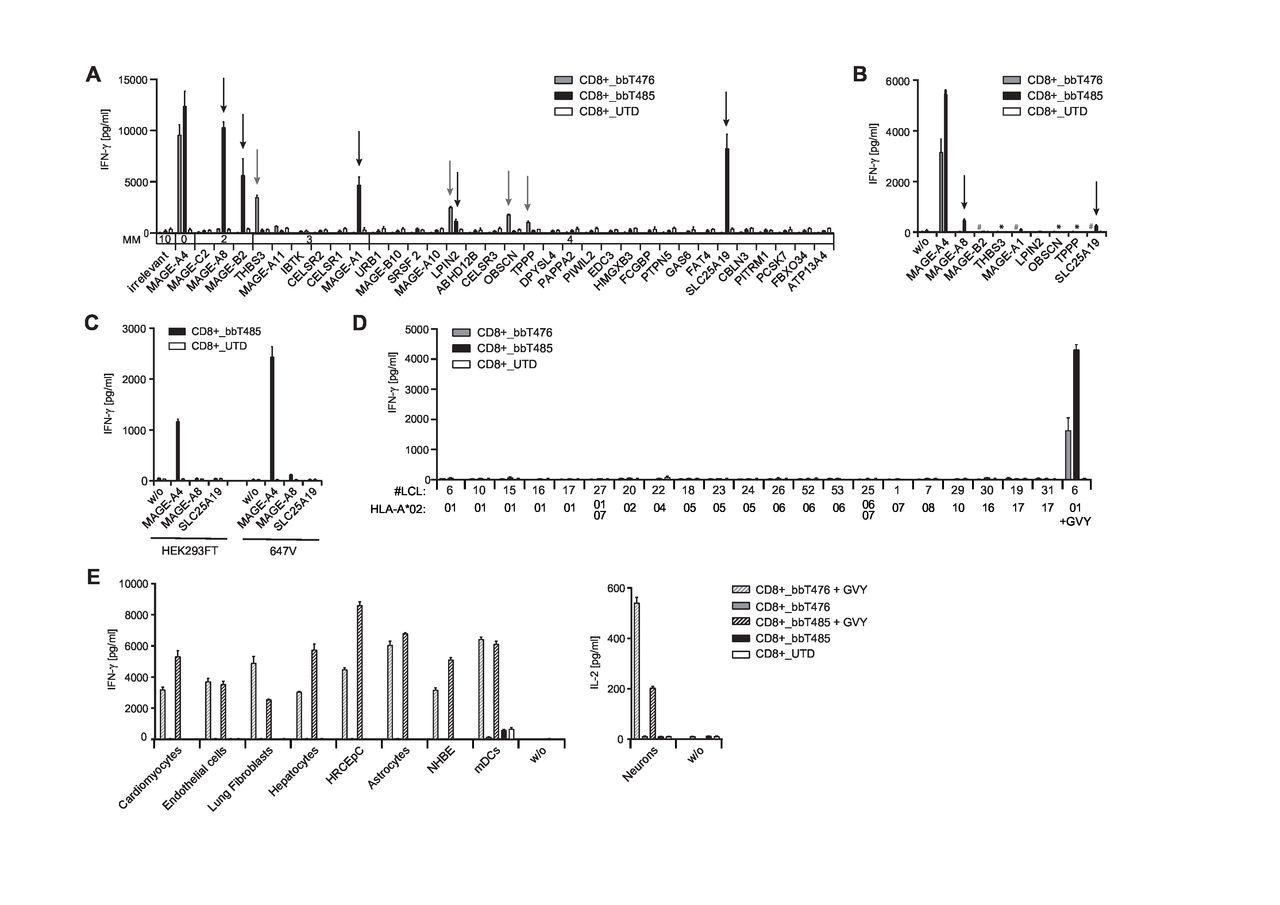
To assess risks for off-target cross-reactivity, TCR-Ts were tested for recognition of peptides partially homologous to the GVY epitope that are likely to be found in the processed “epitome” presented by HLA-A*02:01-encoded molecules. All peptides with up to four mismatches identified by the Expitope web server37 were assessed for their capacity to stimulate cytokine release after loading onto T2 cells. CD8+ T cells of bbT476 and bbT485 cross-recognized four (THBS3, LPIN2, OBSCN, and TPPP) and five (MAGE-A8, MAGE-B2, MAGE-A1, LPIN2, and SLC25A19) mismatched peptides, respectively (figure 3A).
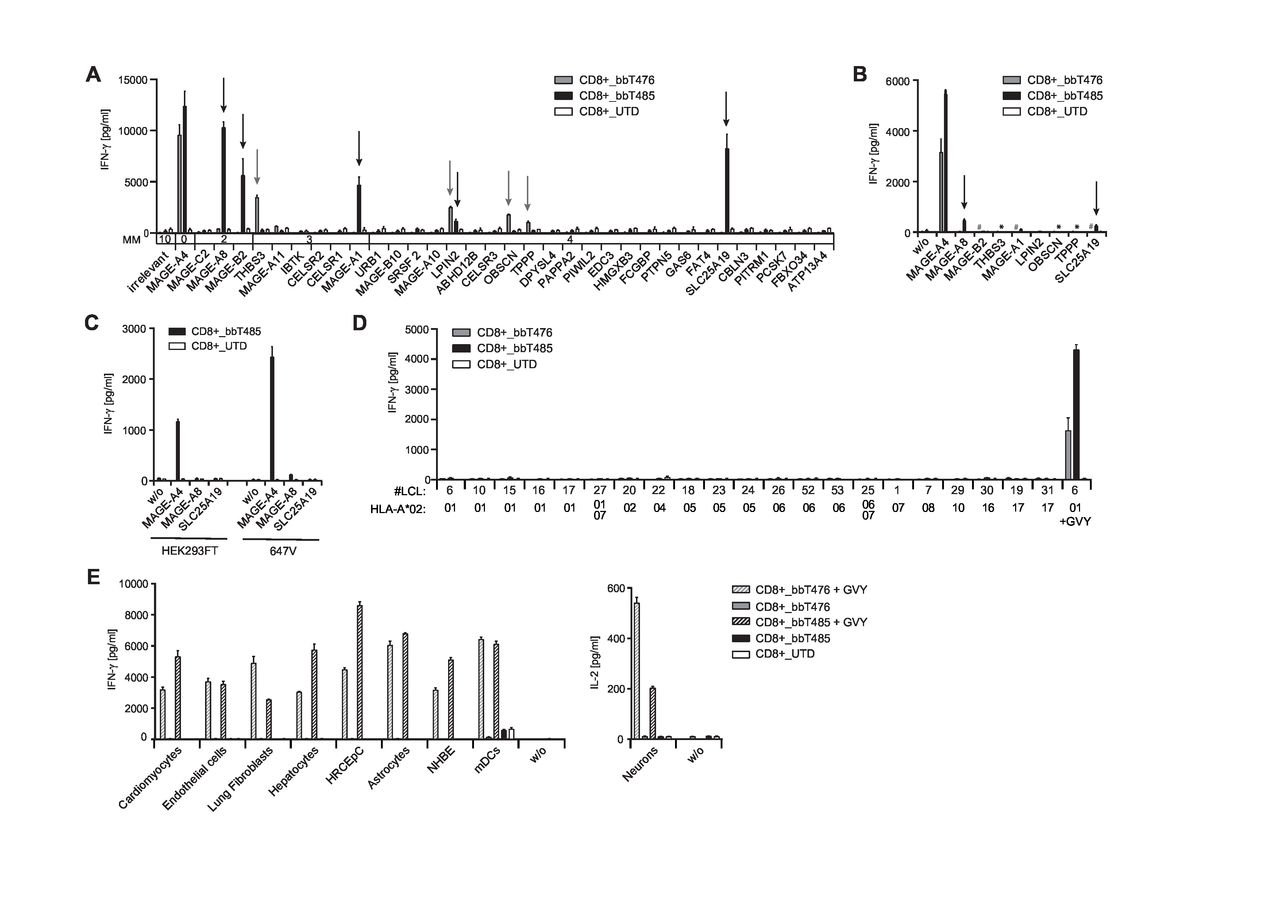
bbT476 and bbT485 TCR-Ts both display excellent safety profiles for adoptive cell therapy. Bar graphs displaying IFN-γ concentrations in supernatants of untransduced (UTD) and TCR-expressing (bbT476 or bbT485) CD8+ T cells 16 hours after co-culture with HLA-A2-positive target cells relevant for safety testing: (A) T2 cells loaded with predicted mismatched peptides using Expitope or GVY peptide. Parental protein names indicate respective mismatched peptide. MM=number of amino acid mismatches compared with GVYDGREHTV. Arrows mark recognized mismatched peptides. (B) A2+K562 cells electroporated with ivt-RNA encoding midi-gene fragments containing the mismatched peptide sequences or GVYDGREHTV (MAGE-A4). Parental protein names indicate transfected midi-genes. Arrows mark recognized transfectants. *bbT485 not tested, #bbT476 not tested. (C) 647V cells and HEK293T cells stably transduced with indicated full-length genes. (D) LCLs expressing different HLA-A*02 sub-alleles as indicated on x-axes. LCL loaded with the GVY peptide serve as positive control. (E) Healthy tissue cells (primary or iPSC-derived) loaded and not loaded with GVY peptide; concentrations for IFN-γ (left panel) or IL-2 (right panel) are depicted. w/o=without target cells. Data shown are representative of 3 different donors for each tested TCR.
To further assess if individual cross-recognized peptides could be processed endogenously from original protein sequences and presented at levels that facilitate recognition by MAGE-A4-reactive TCRs, midi-gene fragments were designed and electroporated in MAGE-A4-negative target cells as ivt-RNA. Translation of ivt-RNA was confirmed using a control TCR recognizing an epitope encoded by a sequence placed at the 3′ end of the ivt-RNA (online supplemental figure 3A). Recognition of the control epitope on A2+K562 cells was on average twofold higher when compared with three other cell lines tested (data not shown). CD8-enriched TCR-Ts with both TCRs showed robust recognition of A2+K562 cells expressing the MAGE-A4 midi-gene. CD8+ cells of bbT476 did not recognize any of the other ivt-RNAs tested (figure 3B) while CD8+ cells of bbT485 showed minimal recognition of A2+K562 cells overexpressing the MAGE-A8-derived and SLC25A19-derived epitopes (figure 3B). To examine near-physiological expression levels, the MAGE-A8 or SLC25A19 genes were stably introduced into MAGE-A4-negative cell lines using a retroviral vector, and transgene expression was confirmed by eGFP-reporter expression (online supplemental figure 3B). CD8+ cells of bbT485 showed strong recognition of MAGE-A4-expressing 647V and HEK293T cells but no recognition of corresponding negative controls (eGFP only). Importantly, no recognition of 647V and HEK293T cells expressing the complete MAGE-A8 or SLC25A19 genes was apparent (figure 3C).
In addition to cross-recognition of mismatched peptides, HLA allo-cross-reactivity of a TCR toward a different HLA allotype could have serious consequences when TCR-Ts are administered to a patient carrying such HLA alleles.38 To assess potential TCR-mediated cross-reactivity against other classical HLA allotypes, a panel of LCLs was tested in co-culture assays with the TCR-Ts. No cross-recognition of HLA-A2 sub-alleles (figure 3D) nor recognition of other common HLA-A, HLA-B, and HLA-C allotypes (online supplemental table 1) was observed for CD8+ T cells expressing either of the two TCRs (online supplemental figure 3C). Recognition of GVY-peptide-loaded LCLs with different HLA-A2 sub-alleles showed a similar recognition pattern for both TCRs (online supplemental figure 3D), indicating their very similar fine specificities.
To further evaluate the risk for off-target reactivity, nine primary human cells and induced pluripotent stem cell (iPSC)–derived cells were assessed as target cells. The HLA-A*02:01-negative normal human bronchial epithelial (NHBE) cells were transfected with ivt-RNA encoding HLA-A*02:01 prior to the co-culture setup. HLA-A2 expression in neurons was induced via IFN-γ pretreatment and IL-2 production was used to measure T-cell activation. HLA-types are given in online supplemental table 2 and expression of HLA-A2 on all cell types was confirmed by flow cytometry (online supplemental figure 4) and by robust recognition by TCR-Ts in co-culture with GVY-peptide-loaded target cells (figure 3E). Notably, CD8+ cells of both TCR-T products did not cross-recognize any healthy cells of the test panel (figure 3E). Altogether, these results demonstrated that TCR-Ts expressing either of the two TCRs had favorable safety profiles.
bbT485 TCR-Ts are more efficacious than bbT476 TCR-Ts in vitro and in vivo
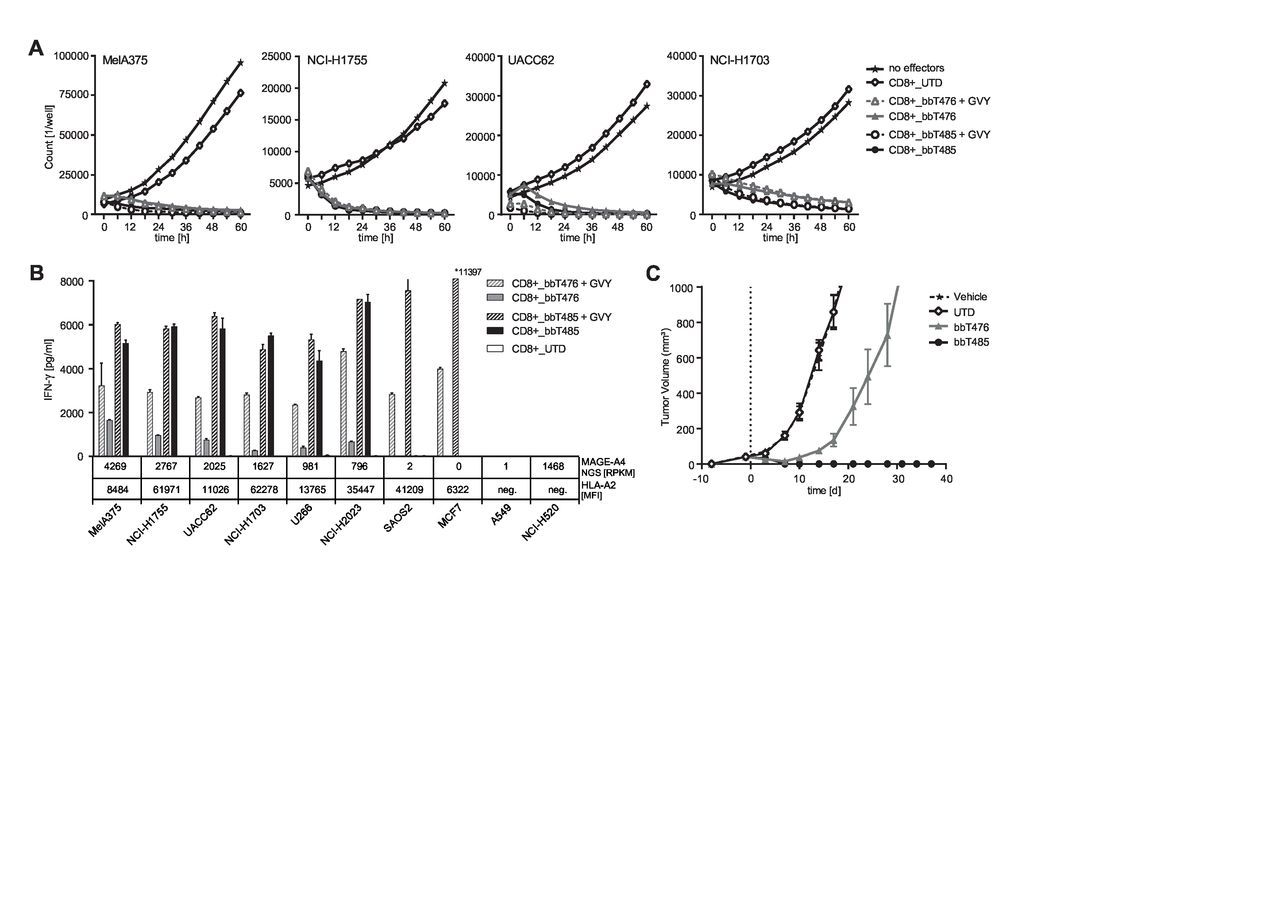
Cytotoxic effects of TCR-Ts were analyzed using the IncuCyte ZOOM-Live Cell Analysis System, which enables direct counting of exclusively viable target cells. Target cells endogenously expressing medium to high levels of MAGE-A4 were rapidly and effectively lysed by both TCR-T products (figure 4A and online supplemental figure 5).

bbT485 TCR-Ts are more efficacious than bbT476 TCR-Ts in vitro and in vivo. (A,B) Reactivity of untransduced (UTD) and TCR-expressing (bbT476 or bbT485) CD8+ T cells toward MAGE-A4-positive tumor cell lines was tested using different readouts. +GVY indicates pulsing of tumor cells with GVY peptide before co-culture assay. (A) Graphs showing the cell number of MAGE-A4-positive tumor cells expressing NucLight Red live-cell labeling reagent (mKate) for real-time imaging in co-culture with T cells over time. No effectors=without T cells. MAGE-A4 expression of tumor cells is ordered from highest (left) to lowest (right). (B) Bar graphs displaying IFN-γ concentrations in supernatants of T cells 16 hours after co-culture with tumor cells. MAGE-A4 NGS [RPKM]=MAGE-A4 RNA level according to TRON Cell Line Portal,58 HLA-A2 [MFI]=HLA-A2 expression level defined by the median fluorescence intensity as evaluated by flow cytometry. HLA-A2 and/or MAGE-A4 negative cell lines serve as negative controls. Data shown are representative of 3 different donors for each tested TCR. (C) Female NOD-Cg-Prkdcscid IL2rgtm1Wjl/SzJ mice received subcutaneous injections of MAGE-A4-positive HLA-A*02:01-positive MelA375 melanoma tumor cells to establish xenografts. At 9 days post tumor implantation, randomized groups of 5 mice each received a single intravenous injection of either media alone or cell suspensions containing TCR-Ts of either bbT476 or bbT485, or an equivalent number of untransduced T cells. Mice were monitored until day 37 post T-cell injection and tumor volume was measured via calipers ~2 times a week and calculated using the formula: length×width×height×π/6.
To further explore potential differences between the two TCRs, IFN-γ cytokine release was assayed with a larger panel of MAGE-A4-positive and MAGE-A4-negative cell lines. The maximum capacity of cytokine release by TCR-Ts is influenced by target cell characteristics such as HLA-A2 expression levels or inhibition mediated by regulatory molecules. Therefore, GVY-peptide-loaded cells served as indicators for the maximum recognition level of each respective tumor cell line by an individual TCR-T-cell product. CD8+ T cells of bbT476 only showed maximal IFN-γ secretion when target cells were loaded with GVY peptide (figure 4B). This trend was not apparent for CD8+ T cells of bbT485, as IFN-γ secretion levels were comparable regardless of whether target cells were peptide-pulsed or not, strongly suggesting superior activity of bbT485 TCR-Ts. MAGE-A4-negative or HLA-A2-negative tumor cell lines were not recognized (figure 4B), demonstrating the exquisite peptide selectivity of both products.
To investigate whether TCR-T products containing both CD8+ and CD4+ T cells could mediate tumor responses in vivo, a murine xenograft model was used for TCR-T treatment of subcutaneous MelA375 solid tumors. After a single intravenous injection, bbT476 TCR-Ts only slowed tumor outgrowth whereas all mice treated with bbT485 TCR-Ts demonstrated complete tumor regression without any signs of relapse measured up to day 37 (figure 4C).
These data indicated that CD8+ T cells expressing both TCRs could effectively lyse tumor cells in vitro, but only bbT485 expressing the allo-derived high-avidity TCR was able to completely eradicate MAGE-A4-positive tumors in vivo.
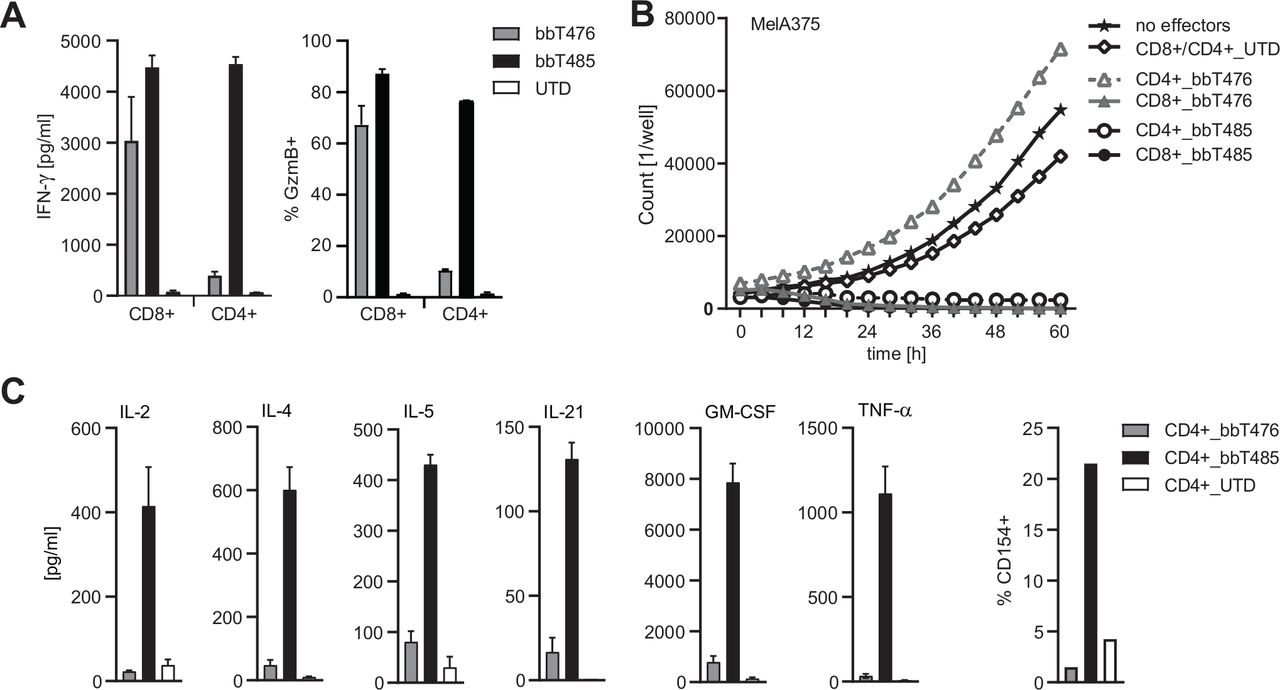
CD8 co-receptor independence of the allo-derived TCR enables a CD4+ T-cell-mediated anti-tumor response
Differences in tumor control were very clear when the two TCR-T products were tested in vivo. Since both CD4+ and CD8+ effector T cells expressing the respective TCR were injected into the tumor-bearing mice, we hypothesized that CD4+ T cells expressing the allo-derived TCR supported superior tumor control by bbT485. It was shown previously that T cells expressing high-affinity HLA class I-restricted TCRs could be activated independently of the CD8 co-receptor, thereby transferring desired effector functions to CD4+ T cells.39 To investigate co-receptor dependency, CD4+ T cells were tested in co-cultures with MAGE-A4-expressing MelA375 cells. Only CD4+ T cells of bbT485 exhibited hallmarks of activation and cytotoxicity, as determined by IFN-γ secretion and Granzyme B expression (figure 5A), respectively. Furthermore, TCR-Ts with the autologous-derived TCR demonstrated target cell lysis only when expressed in CD8+ T cells. However, CD4+ TCR-Ts expressing the allo-derived TCR were able to kill MAGE-A4-positive target cells, highlighting the co-receptor-independent signature and potency of the allo-derived TCR (figure 5B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Co-receptor independence of the allo-derived TCR enables CD4+ T-cell-mediated anti-tumor response. Graphs showing different read-outs of activation of CD8+ and CD4+ T cells expressing transgenic TCRs (bbT476 or bbT485) or not (UTD, untransduced) in co-culture with MAGE-A4-positive HLA-A*02:01-positive MelA375 cells. (A) Bar graphs displaying IFN-γ concentrations in supernatants (left panel) and Granzyme B (GzmB) expression among CD8+ and CD4+ T cells determined by flow cytometry (right panel) 16 hours after co-culture. (B) Graph showing the cell number of MelA375 cells expressing NucLight Red live-cell labeling reagent (mKate) for real-time imaging in co-culture with T cells. No effectors=without T cells. (C) Quantification of cytokine levels in supernatants (left panel) and CD154 expression among CD4+ T cells determined by flow cytometry (right panel) 16 hours and 6 hours after co-culture, respectively. Data shown are representative of 3 different donors for each tested TCR.
Beyond direct anti-tumor effects, CD4+ T cells can also display polyfunctional cytokine responses that could contribute to tumor control in vivo. CD4+ T cells of bbT485 secreted a variety of different cytokines, including IL-2, IL-4, IL-5, IL-21, GM-CSF, and TNF-α after antigen-specific activation (figure 5C). In addition, only CD4+ TCR-Ts of bbT485 upregulated CD154 surface expression (figure 5C).
These data demonstrated that, in addition to its superior activity profile, the allo-derived TCR is CD8 co-receptor independent, thereby allowing CD4+ TCR-Ts to support anti-tumor reactivity through direct tumor cell killing and polyfunctional cytokine secretion, both of which may explain the superior tumor control in vivo for bbT485 TCR-Ts.
Discussion
Adoptive transfer of T cells with redirected specificities has dramatically improved treatment options for selected cancer types, such as treatment of B-cell malignancies with CD19-specific CAR-Ts.40 41 For many other cancer indications, the choice of a suitable antigen is much more challenging. The most promising group of TAAs studied to date are cancer-testis antigens as their expression in healthy cells is often limited to the immune-privileged testis tissue.8 9 The prominent expression of MAGE-A4 in several solid tumor indications makes it a promising target for adoptive cell therapy with TCR-Ts. However, MAGE-A4 expression in metastases remains to be studied and patient selection needs to be adapted accordingly.
A first challenge in targeting an antigen with a TCR is to identify immunogenic peptides that are efficiently processed and presented on a given HLA molecule and can be recognized by T cells. Bioinformatics tools using algorithms to predict the affinity to HLA and especially the immunogenicity of epitopes are often inaccurate. Therefore, we functionally screened for MAGE-A4-derived peptides that are processed, presented, and recognized by T cells. The identified MAGE-A4-derived peptide (GVYDGREHTV) would not have been chosen based only on prediction algorithms due to its low predicted affinity for HLA-A*02:01-encoded molecules.34 35
The isolation of high-affinity TCRs binding to peptides derived from cancer-testis antigens is limited due to their expression in the thymus42 and the resulting negative selection of respective T cells. To circumvent the limitations of tolerant T-cell repertoires, we used an allogeneic priming approach previously shown to yield excellent TCRs specific for different tumor-associated antigens.19 43 44
MAGE-A4 expression in non-malignant healthy tissues is restricted to immune-privileged sites, reducing the risk of on-target, off-tumor toxicity to very low levels.8 9 45 However, unknown off-target cross-reactivity can lead to severe adverse events for patients.16 46 47 Due to major differences in murine and human proteomes, mouse models are not suited for in vivo toxicology studies and therefore robust in vitro experiments are required to investigate potential off-target toxicities. Partially homologous peptides that were cross-recognized when loaded on target cells at high concentrations could be successfully de-risked for both TCR-Ts when assessed under more physiological conditions. Furthermore, off-target reactivities were not observed when a panel of normal cells representing vital organs or HLA-A2-positive MAGE-A4-negative tumor lines and LCLs were tested for TCR-T recognition, confirming the peptide selectivity of both TCRs.
Another source of off-target toxicity could derive from recognition of HLA molecules independent of recognition of a defined peptide since the healthy donors from which the TCRs were isolated do not share all HLA allotypes with TCR-Ts that will be used to treat different patients.38 No such cross-reactivity was observed in a panel of LCLs expressing the most common HLA allotypes. Although in vitro assays can never completely predict the behavior of TCR-Ts in patients, none of the performed analyses gave any indication of potential off-target toxicity.
The superior efficacy of TCR-Ts expressing the allo-derived TCR was seen when cytokine release after recognition of tumor cells was analyzed and even more pronounced pre-clinical efficacy was observed in an in vivo xenograft model. We hypothesized that the superior in vivo efficacy could be due to the ability of the TCR-Ts to function independently of CD8 co-receptors on CD4+ T cells. Injection of CD4+ and CD8+ T cells targeting the same antigen can produce synergistic effects that lead to complete eradication of malignant cells.48 Target antigen-specific CD4+ T cells can execute direct effector functions and also support the proliferation and survival of CD8+ T cells, for example, by the release of cytokines like IL-2. In addition, the production of IL-21 by CD4+ T cells was shown recently to be essential for formation of durable CD8+ T-cell responses during chronic antigen exposure.49 50 Furthermore, antigen-presenting cells can be activated by CD154 expressed on CD4+ T cells, enhancing and widening the immune response at different levels.51 Therefore, antigen-specific CD4+ T cells when added to antigen-specific CD8+ T-cell products may increase treatment efficacy, especially relevant for solid tumors.
For TCRs, there are several options to generate antigen-reactive CD4+ and CD8+ T cells. First, the two T-cell populations could be modified with two different TCRs, one that is HLA class I restricted and functions on CD8+ cells and one that is HLA class II restricted and functions on CD4+ cells. However, this would dramatically increase costs and efforts for development and clinical testing. In addition, HLA class II molecules are often poorly expressed on tumor cells. Second, the CD8 co-receptor can be transferred in parallel with a CD8-dependent TCR, enabling the TCR to also function on CD4+ T cells.52 As sequence space in vectors is often limited, the addition of CD8 would narrow the possibility to transfer additional modules like chimeric receptors53–55 and cytokines that might enhance efficacy in the tumor microenvironment, or safety switches.56 Third, a CD8-independent TCR that functions on CD4+ T cells without additional CD8 can be transferred to CD8+ and CD4+ T cells in parallel, having the clear advantage that the same vector can be used for both T-cell populations and that vector space remains available for additional sequences.
Conclusion
Our extensive in vitro and in vivo studies demonstrated that we successfully isolated a MAGE-A4-specific high-avidity TCR with an excellent safety profile using an allo-restricted priming approach. This TCR shows superior in vitro and in vivo killing capacities when compared with TCR-Ts expressing a TCR derived from autologous (tolerized) T cells. Importantly, this MAGE-A4-specific TCR displayed CD8 co-receptor independence, giving it the potential to effectively function in either CD4+ or CD8+ T cells, a feature that goes beyond what is currently tested in the clinic.57 Thus, mixed TCR-T populations can be developed for clinical studies in HLA-A*02:01-positive patients. Based on our results, using TCR-T populations including both CD8+ and CD4+ T-cell subsets will have clear advantages for patients bearing solid tumors expressing the MAGE-A4 antigen.
Acknowledgments
We thank the whole TCR Platform, Translational Medicine, and Automation team at Medigene and especially Lisa Siwig, Bettina Bauer, and Marina Hereth for technical assistance and performing experiments for the manuscript, and Gary Wanders and Dominik ter Meer for stimulating discussions. We also thank Andrew Chavkin, Robert Chain, and Thomas Giordano from bluebird bio for manufacturing of transgenic TCR-T-cells used in studies.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors KD, TH, DS, and CE conceived and designed the work. KD, TH, and LP acquired and analyzed most of the data. GL established and advised the CD137 sorting method. KPG and LJP conducted experiments, acquired data, and analyzed data. ID and PRN designed experiments and analyzed data. MSM designed the experiments, analyzed data, and revised the manuscript. KD, TH, and DS drafted the manuscript, which was critically revised by DJS and CE for important intellectual content. All authors approved the final version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. KD, DS, and CE had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. KD and TH are joint first authors and DS and CE are joint senior authors. Order of co–first and senior authors was decided by consensus.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests KD, TH, LP, GL, DJS, DS, and CE are employees and DJS is a Managing Director of Medigene Immunotherapies GmbH, a subsidiary of Medigene AG, Planegg, Germany. CE and DS are designated as inventors on two patent applications (PCT/EP2020/058779 and PCT/US20/31796) related to this work that have been filed by Medigene Immunotherapies GmbH and bluebird bio Inc. KPG, LJP, PRN, and MSM are current employees at bluebird bio Inc., Cambridge, USA. ID was an employee of bluebird bio., Cambridge, USA during her contributions to this publication. ID is a current employee of ElevateBio, Waltham, USA. KPG, LJP, ID, PRN, and MSM are current equity holders at bluebird bio., Cambridge, USA.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Data availability will be possible from KD.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.