Article Text

Abstract
Background High serum interleukin (IL-6) levels may cause resistance to immunotherapy by modulation of myeloid cells in the tumor microenvironment. IL-6 signaling blockade is tested in cancer, but as this inflammatory cytokine has pleiotropic effects, this treatment is not always effective.
Methods IL-6 and IL-6R blockade was applied in an IL-6-mediated immunotherapy-resistant TC-1 tumor model (TC-1.IL-6) and immunotherapy-sensitive TC-1.control. Effects on therapeutic vaccination-induced tumor regression, recurrence and survival as well on T cells and myeloid cells in the tumor microenvironment were studied. The effects of IL-6 signaling in macrophages under therapy conditions were studied in Il6rafl/fl×LysMcre+ mice.
Results Our therapeutic vaccination protocol elicits a strong tumor-specific CD8+ T-cell response, leading to enhanced intratumoral T-cell infiltration and recruitment of tumoricidal macrophages. Blockade of IL-6 signaling exacerbated tumor outgrowth, reflected by fewer complete regressions and more recurrences after therapeutic vaccination, especially in TC-1.IL-6 tumor-bearing mice. Early IL-6 signaling blockade partly inhibited the development of the vaccine-induced CD8+ T-cell response. However, the main mechanism was the malfunction of macrophages during therapy-induced tumor regression. Therapy efficacy was impaired in Il6rafl/fl×LysMcre+ but not cre-negative control mice, while no differences in the vaccine-induced CD8+ T-cell response were found between these mice. IL-6 signaling blockade resulted in decreased expression of suppressor of cytokine signaling 3, essential for effective M1-type function in macrophages, and increased expression of the phagocytic checkpoint molecule signal-regulatory protein alpha by macrophages.
Conclusion IL-6 signaling is critical for macrophage function under circumstances of immunotherapy-induced tumor tissue destruction, in line with the acute inflammatory functions of IL-6 signaling described in infections.
- adaptive immunity
- immunotherapy
- active
- macrophages
- CD8-Positive T-Lymphocytes
- immune evation
Data availability statement
Data are available on reasonable request. The TC-1.control and TC-1.IL-6 cell lines are available on request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
High serum concentrations of interleukin (IL-6) are detected in patients with large tumors or advanced disease stage and correlates with worse performance status and resistance to chemotherapy.1 2 IL-6 is produced by tumor cells, various immune and non-immune cells and has a wide range of effects in hematopoiesis, immune defense and oncogenesis.3 IL-6 stimulates the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway, predominantly activating STAT1 and STAT3. To prevent pathophysiological consequences of IL-6 signaling, this pathway is under tight control with a prominent role for suppressor of cytokine signaling 3 (SOCS3) to inhibit JAK-STAT3 activation and targeting cytokine receptor complexes for degradation.3 Normal physiological serum concentrations of IL-6 are in the picogram per ml range but serum levels increase to micrograms per ml during different disease settings.4
IL-6 may stimulate tumor cell proliferation and induce tumor resistance to chemotherapy in vitro, by the activation of several intracellular signaling pathways, upregulation of multidrug resistance genes and antiapoptotic proteins or by reduced caspase-3 activation,3 5–8 all of which can stimulate cancer cell growth. Tumor-produced IL-6 can also alter the tumor microenvironment by promoting chronic inflammation and suppressing antitumor immunity. Specifically, it may impair the induction and tumor infiltration of type 1 T cells while stimulating the attraction of myeloid derived suppressor cells and Tregs.7 9 IL-6 also suppresses the antigen presenting function of dendritic cells (DCs) and macrophages and is involved in the switch of tumor-associated macrophages from a M1 phenotype to a M2 phenotype.7 9–12 Antibody-mediated blockade of IL-6 effectively slows down the outgrowth of some tumors6 12 coinciding with restoration of T-cell priming and migration.12 In other models, IL-6 blockade requires cotreatment with PD-L1 blockade7 11 or transforming growth factor (TGFβ) blockade13 to become effective.
We recently demonstrated that TC-1 tumor cells engineered to produce IL-6 were resistant in vivo to chemotherapy as well as immunotherapy in the form of a therapeutic vaccine. Whereas wild-type tumors all fully regressed due to the concerted action of vaccine-induced tumor-specific T cells and macrophages,14 the IL-6 producing tumors quickly recurred after an initial regression phase.10 This was not caused by a changed in vitro or in vivo growth rate of tumor cells, a change in sensitivity to different (T-cell mediated) cytotoxic attacks, an impaired vaccine-mediated induction of tumor-specific T cells or the infiltration of T cells into tumors. Instead, IL-6-mediated resistance was associated with the functional inhibition of several subsets of myeloid cells.10
In the current study, we tested if blockade of IL-6 signaling in this tumor model would revert the immunosuppressive activity on the stromal components. Surprisingly, we observed that blockade of IL-6 signaling during immunotherapy aggravated disease, reflected by an impaired tumor regression and an increased recurrence rate. This was primarily due to loss of IL-6 proinflammatory signaling in tumor-resident macrophages and associated with a decreased SOCS3 and an increased signal-regulatory protein alpha (SIRPα) expression by these myeloid cells. Our data show that IL-6 blockade under circumstances where the immune system actively causes tissue destruction and results in significant tumor regression may be harmful, most likely because it interferes with those functional activities of T cells and macrophages that are required during more acute inflammatory processes (eg, infections).3 15
Materials and methods
Mice
Wild-type female C57BL/6 mice aged 6–were obtained from Charles River Laboratories. Il6rafl/fl×LysMcre- and Il6rafl/fl×LysMcre+ transgenic mice were obtained from University Medical Center Hamburg-Eppendorf, (UKE, Germany) and were generated as previously described.16 17 Mice were housed in individually ventilated cages under specific pathogen-free conditions in the animal facility of Leiden University Medical Center (LUMC, Leiden, the Netherlands).
Tumor cell line and culture conditions
The tumor cell line TC-1 was generated by retroviral transduction of C57BL/6 lung epithelial cells with the HPV16 E6/E7 and c-H-ras oncogenes18 and cultured as described previously.19 TC-1.control and IL-6 tumor cell lines were made by transfection of TC-1 tumor line with pcDNA3.1 vector with hygromycin resistance gene. This vector is adapted by replacement of CMV promotor with a short elongation factor (EFS) promotor. A fragment was inserted with the IRES sequence and GFP. IL-6 gene was inserted between EFS promotor and IRES sequence and GFP (TC-1.IL-6). For a control, no gene was inserted between EFS promotor and IRES sequence and GFP (TC-1.control). These cell lines were reported previously.10 All the cell lines were cultured in Iscove’s Modified Dulbecco’s Media (IMDM) (BioWhittaker) supplemented with 8% fetal calf serum (FCS) (Greiner), 2 mM L-glutamine (Life Technologies), 50 IU/mL penicillin (Life Technologies) and 50 µg/mL streptomycin (Life Technologies). Cells were cultured in a humidified incubator at 37°C and 5% CO2. Mycoplasma tests that were frequently performed for all cell lines by PCR were negative. All experiments were performed with mycoplasma-free cells.
Tumor experiments and treatments
Mice were inoculated subcutaneously with 1×105 tumor cells in 200 µL PBS containing 0.2% BSA on day 0. Tumor size (horizontal dimension×vertical dimension) was measured two times a week using a caliper. When a palpable tumor was present (day 8), mice were divided into groups with comparable tumor sizes. On day 8 post-tumor challenge, mice were treated with synthetic long peptide (SLP) vaccine. The SLP vaccine containing 100 µg human papilloma virus type 16 (HPV16) E743-63 (GQAEPDRAHYNIVTFCCKCDS) with 20 µg CpG (ODN1826, InvivoGen) dissolved in 50 µL PBS was administrated subcutaneously in tail base of mice. For tumor outgrowth and survival experiments, boost vaccine was given on day 22 post-tumor challenge (online supplemental figure S1A). Rat anti-mouse IL-6R antibody (clone: MR16-1, a kind gift from Chugai Pharmaceutical, Tokyo, Japan) was applied to block the IL-6R. To block IL-6 in vivo, Mab antimouse IL-6 (clone: MP5-20F3, BioXcell) was used. Both antibodies were given at 200 µg per mouse dissolved in PBS in 200 µL and administrated intravenously in the tail vein or retro-orbital every 3 days for total seven injections. For local injection of anti-IL-6R, 200 µg of the antibody per mouse dissolved in PBS in 50 µL and administrated subcutaneously next to the tumor. These antibodies were administered from 8 to 29 (tumor outgrowth and survival experiments) or 8 to 15 (tumor analysis experiments) post-tumor challenge unless stated differently in the legend of figures. To deplete granulocytic myeloid cells anti-Ly6G (clone 1A8, BioXCell) antibody treatment was started on day 14 (100 µg/mouse) and repeated every 2–3 days (50 µg/mouse) till day 27. Therapy response rates according to RECIST criteria; NR: no response, PR: partial response, defined as a decrease in tumor size, at least two consecutive measurements and minimal 30% of lesion, CR: complete response, defined as a complete disappearance of a previous tumor. For tumor analysis experiment, mice were sacrificed on day 16 post tumor challenged (online supplemental figure S1A).
Supplemental material
Flow cytometric analysis of splenic and tumor-infiltrating immune cells
For analysis of (tumor-infiltrating) immune populations, tumors were disrupted in small pieces and incubated with 0.4 mL/mL Liberase TL Research grade (Roche) in IMDM for 15 min at 37°C. Spleens were digested by incubating with 0.02 mg/mL DNAse (Deoxyribonuclease I from bovine pancreas, Sigma-Aldrich) and 1 mg/mL collagenase D (Roche) for 10 min at room temperature. Single-cell suspensions were prepared by mincing spleen and tumor pieces through a 70 µm cell strainer (BD Biosciences). Cells were resuspended in staining buffer (PBS+2% FCS+0.05% sodium azide) or brilliant stain buffer (BD Biosciences) and incubated with various fluorescently labeled antibodies against: CD40 (clone 3/23), CD86 (clone PO3 or GL-1), CD70 (clone FR70), CD274 (clone MIH5), CD62L (clone MEL-14), CD163 (clone S15049I), CD103 (clone 2E7), XCR1 (clone ZET), Siglec-H (clone 440c), Siglec-F (clone E50-2440), CCR2 (clone SA203G11 or 440c), CD8a (clone 53–6.7), CD3 (clone 145–2 C11), CD19 (clone 1D3), NK1.1 (clone PK136), CD11b (clone M1/70), CD11c (clone N418 or HL3), CD64 (clone X54-5/7.1), SIRP-α (clone P84), CD45 (clone 30F11), CD45.2 (clone 104), Egr2 (clone erongr2), Arg1 (clone A1exF5), iNOS (clone CXNFT), F4/80 (clone BM8), Ly6C (clone HK1.4), Ly6G (clone 1A8), class II (clone M5/114.15.2) IL-6R (clone D7715A7) and IL-6 (clone MP5-20F3). Antibodies were obtained from BD Biosciences, eBioscience and Biolegend. APC-labeled H-2Db tetramers containing HPV16 E749-57 peptide (RAHYNIVTF) were used as E7 tetramer (E7 Tm). For dead cell exclusion, 7-aminoactinomycin D (7-AAD; Invitrogen), Zombie UV Fixable Viability Kit (Biolegend) or Zombie Aqua or NIR Fixable Viability Kit (Biolegend) were used. For intracellular interferon γ (IFNγ) (clone XMG1.2; BDBiosciences), TNF (clone MP6-XT22; Biolegend), IL-2 (JES6-5H4; Thermofisher), IL-17A (TC11-18H10.1; Biolegend)) and IL-10 (JRS5-16E3; Thermofisher) cytokine staining, single cell suspensions of spleens or tumors were plated in 96-well cell culture flat-bottom plates in the presence of DCs preloaded with SLP and brefeldin A (4 µg/mL). After 5 hours incubation, cells were stained for surface markers and were fixed in fixation buffer (Biolegend) for overnight. For intracellular staining of Egr2, iNOS and Arg1, following surface marker staining, cells were fixed and permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and stained for intracellular markers in Permeabilization Buffer. To measure the production of IL-6 by tumor cells, tumor cells were plated in 96-well cell culture flat-bottom plates in the presence brefeldin A (4 mg/mL). After overnight incubation, cells were fixed in fixation buffer (Biolegend) for 30 min. Thereafter, cells were washed, stained for IL-6. To determine the level of STAT1 and STAT3 on tumor infiltrating cells, cells were fixed with 4% paraformaldehyde and permeabilized with prechilled True-Phos Perm Buffer (Biolegend). Cells were incubated at −20°C for 1 hour. Next, cells were washed and stained with PE anti-STAT1 Phospho (Ser727) (clone A15158B, Biolegend) and Alexa Fluor 647 Mouse Anti-Stat3 (pY705) (clone 4/P-STAT3, BD Bioscience) antibodies in staining buffer and incubated for 30 min. To measure SOCS1 and SOCS3 on tumor digested cells, primary goat anti-SOCS1 antibody (EB05040 Everest Biotech) and primary rabbit polyclonal anti-SOCS3 antibody (ab16030 Abacm) were used, respectively. For secondary antibodies, alexa fluor 488 polyclonal donkey anti-goat (H+L) cross-adsorbed secondary antibody (A-11055 ThermoFisher Scientific) and alexa fluor 647 donkey antirabbit IgG (H+L) (A31573, Life Technologies) were used, respectively. Samples were analyzed with a BD LSRII or LSRFortessa or a Cytek Aurora 5-laser spectral flow cytometers, and results were analyzed using FlowJo software (Tree Star).
IL-6 ELISA
Mouse IL-6 DuoSet ELISA and Mouse IL-6R alpha DuoSet ELISA (R&DSystems) and were used to measure the amount of IL-6 and IL-6R. Serum of the mice or supernatant of cultured cells were obtained and proceed as described in the protocol of the kit.
Statistical analysis
Survival for differentially treated mice was compared using the Kaplan-Meier method and the log-rank (Mantel-Cox) test. Additional statistical methods are stated in the legends. All p values <0.05 were considered significant.
Results
IL-6 pathway blockade impairs the efficacy of a therapeutic cancer vaccine
We showed previously that TC-1 tumor cells producing high levels of IL-6 (TC-1.IL-6) were resistant to therapeutic vaccination whereas mice challenged with TC-1.control tumors, releasing low IL-6 levels (figure 1A) were completely cured.10 To overcome this problem, we blocked the IL-6:IL-6R axis from day 8 onwards by using either anti-IL-6 or anti-IL-6R blocking antibody during therapeutic peptide vaccination. The effect of IL-6R blockade is indicated by the rise in serum levels of IL-6 and the soluble IL6 receptor, which because of the blockade can’t be consumed (figure 1B and online supplemental figure S1A,B). However, blockade of the IL-6 axis resulted in ten percent more tumor recurrence (100% vs 89% recurrences, p=0.0837, Fisher’s exact test, two sided) in the parental TC-1.control model after the initial therapeutic vaccination-induced regression phase (figure 1C). In the TC-1.IL-6 model, this blockade more dramatically aggravated clinical response to therapeutic vaccination (figure 1D). The degree of tumor regression was significantly less with many of the treated mice showing only partial or NR (CR+PR vs NR p=0.0022) (figure 1E), resulting in recurrences in almost all of the mice and a worse overall survival (figure 1F and online supplemental figure S1C). Similar observations were made when IL-6 signaling was blocked very early, from day 0 onward, or when blocking antibodies were injected locally to allow sufficient amounts of antibody to drain to the tumor (figure 1D and F). IL-6R blockade displayed only a minor effect on the growth of untreated tumors (online supplemental figure S1D) Thus, blockade of IL-6 signaling impairs the efficacy of T-cell based immunotherapy and this is effect is more evident in the TC-1.IL-6 model.
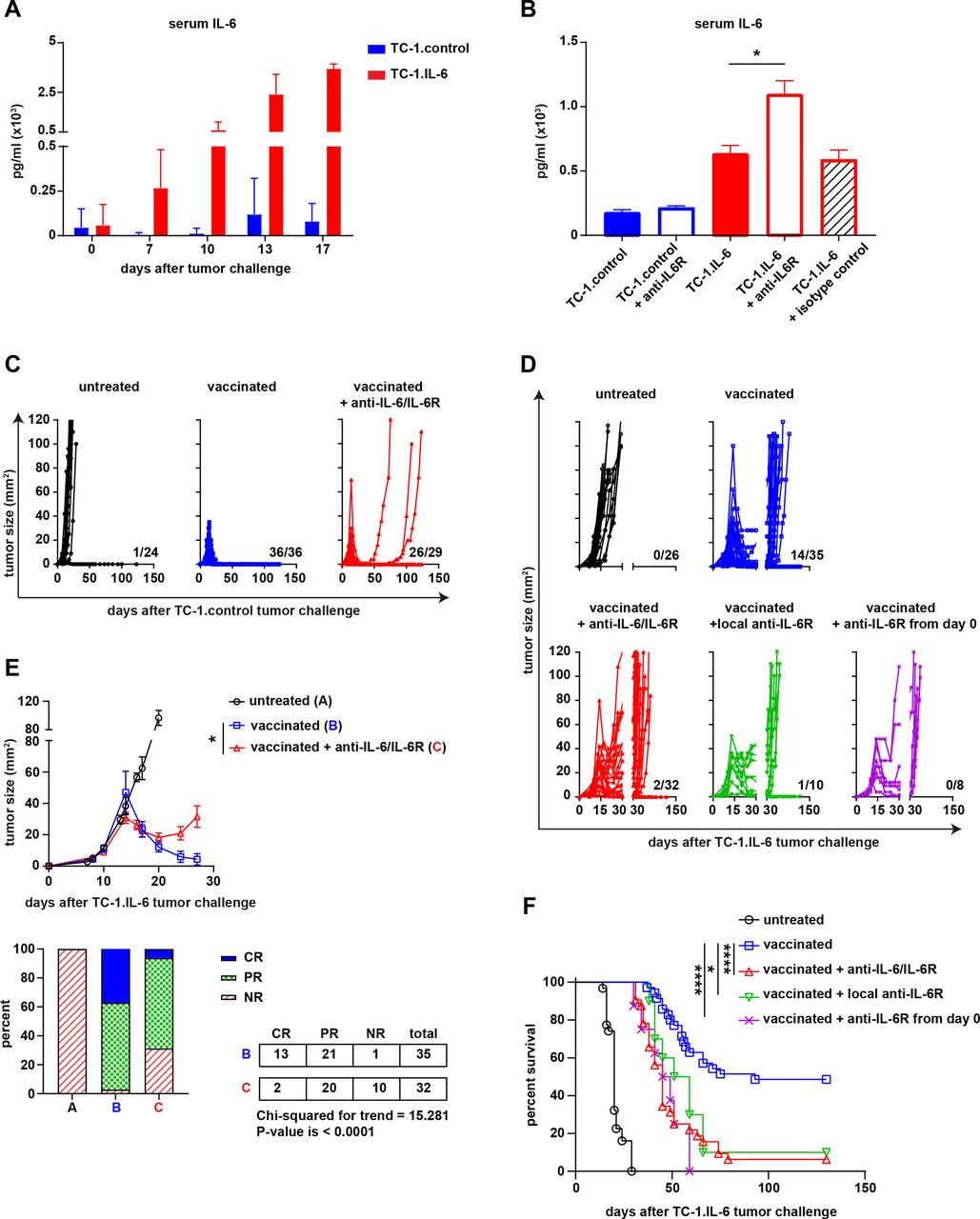
IL-6 blockade impairs the therapeutic efficacy of a therapeutic cancer vaccine. (A) The serum level of IL-6 (pg/mL) in TC-1.contol and TC-1 IL-6 tumor-bearing mice at different time points after tumor challenge. (B) The serum level of IL-6 (pg/mL) in TC-1.contol and TC-1 IL-6 tumor-bearig mice with and without IL-6R blockade or isotype control at day 16 post-tumor challenge. Tumor outgrowth graphs of the untreated or SLP vaccinated TC-1.control (C) and TC-1.IL-6 (D) tumor-bearing mice treated with or without IL-6 or IL-6R antibodies as described in the material and methods. The number shown above the x-axis is the number of alive mice from the total. (E) The average tumor outgrowth (upper panel) and the mean response rate (%) according to RECIST criteria (upper panel) of the major groups shown in (D). Lower panel: NR, no response; PR, partial response; CR, complete response. Graphs indicate mean values with SEM (F) survival graph of the mice shown in (D). Data in (A, B) is representative of three independent experiments, yielding similar results. Data shown in C–F are pooled from four independent experiments with similar results. Significance was determined by Mann-Whitney U test in B and student unpaired t-test for difference in endpoint tumor size in the vaccinated groups and χ2 for trend for response rate in (E). Significance was determined by a log-rank (Mantel–Cox) test in (F). *P<0.05; **p<0.01; ***p<0.001. IL-6, interleukin 6; SLP, synthetic long peptide.
The IL-6-driven HPV-specific T cell response is partially impaired by IL-6 axis blockade
Therapeutic vaccination in the TC-1 tumor model increases the percentage of intratumoral CD45+ immune cells, in particular that of CD8+ and CD11b+ immune cell populations.14 Blocking IL-6/IL-6R axis did not alter the infiltration of CD45+ immune cells (figure 2A, additional file 2), nor the percentage of CD11b+, CD4+ and Tregs within these immune cells in TC-1.control or TC-1.IL-6 tumors (figure 2B). However, IL-6/IL-6R blockade did decrease the percentage of intratumoral CD8+ T cells in TC-1.IL-6 tumor-bearing mice (figure 2B) including HPV-specific CD8+ T cells (figure 2C). This effect was more pronounced in IL-6 producing tumors. IL-6/IL-6R blockade did not alter the immune composition of untreated TC-1.IL-6 tumors (online supplemental figure S3). During progressive tumor growth IL-6 may suppress T cell immunity11 12 but under other conditions, including vaccination, IL-6 has shown to be important for the expansion and migration of effector T cells,20–23 suggesting that the effects on tumor-specific T cells observed in TC-1.IL-6 may be due to a blockade of these processes. Therefore, we analyzed the T-cell response in the blood at different time points (online supplemental figure S2D). One week after vaccination when the tumor regressed (day 16–20), the HPV-specific CD8+ T-cell response was significantly lower when IL-6R blocking antibodies were injected in both models (figure 2D), but after recurrence (day 30) the percentage of HPV-specific T cells was similar and higher, irrespective of IL-6R blockade (figure 2D). There was no contribution of the IL-6 produced by tumor cells to this process of T cell expansion as vaccinated TC-1.control and TC-1.IL-6 tumor-bearing mice show similar E7-specific CD8+ T-cell responses at day 16–20 and 30 (figure 2D). Furthermore, no differences were observed in the SLP vaccination induced increase in percentage of proliferating or effector phenotype of CD8+ T cells after IL-6R blockade (online supplemental figure S4A-D). Also the percentage of vaccine-induced proliferating CD4+ T cells or CD127+KLRG-1+CD4+ T cells or activated CD62L-CD44+CD4+ T cells was not different between the two tumor models (online supplemental figure S4E-H), although that after booster vaccination (day 22), the percentage of activated CD62L-CD44+CD4+ T cells was decreased by IL-6R blockade in TC-1.IL-6 tumor-bearing mice (online supplemental figure S4F).

IL-6 axis blockade partially impairs the tumor-specific T cell response. The percentage of intratumoral CD45+ leukocytes within live cells (A), the mean percentage of CD8+, CD4+ (without Tregs), Tregs and CD11b+ cells within CD45+ cells (B) and the percentage of E7 HPV Tm+ cells within CD45+ cells (C) in untreated and SLP vaccinated TC-1.control and TC-1.IL-6 tumor-bearing mice. (D) The percentage of E7 HPV Tm+ cells within CD8+ T cells in blood on day 16–20 and 30 post-tumor challenge in SLP vaccinated TC-1.control and TC-1.IL-6 tumor-bearing mice with and without IL-6R blockade. Each dot represents data from an individual mouse. Graphs indicate mean values with SEM data are pooled from two independent experiments with similar results. Significance between the vaccinated groups in each tumor model was determined by unpaired Student’s t-test. *P<0.05; **p<0.01. IL-6, interleukin 6; ns, not significant; SLP, synthetic long peptide.
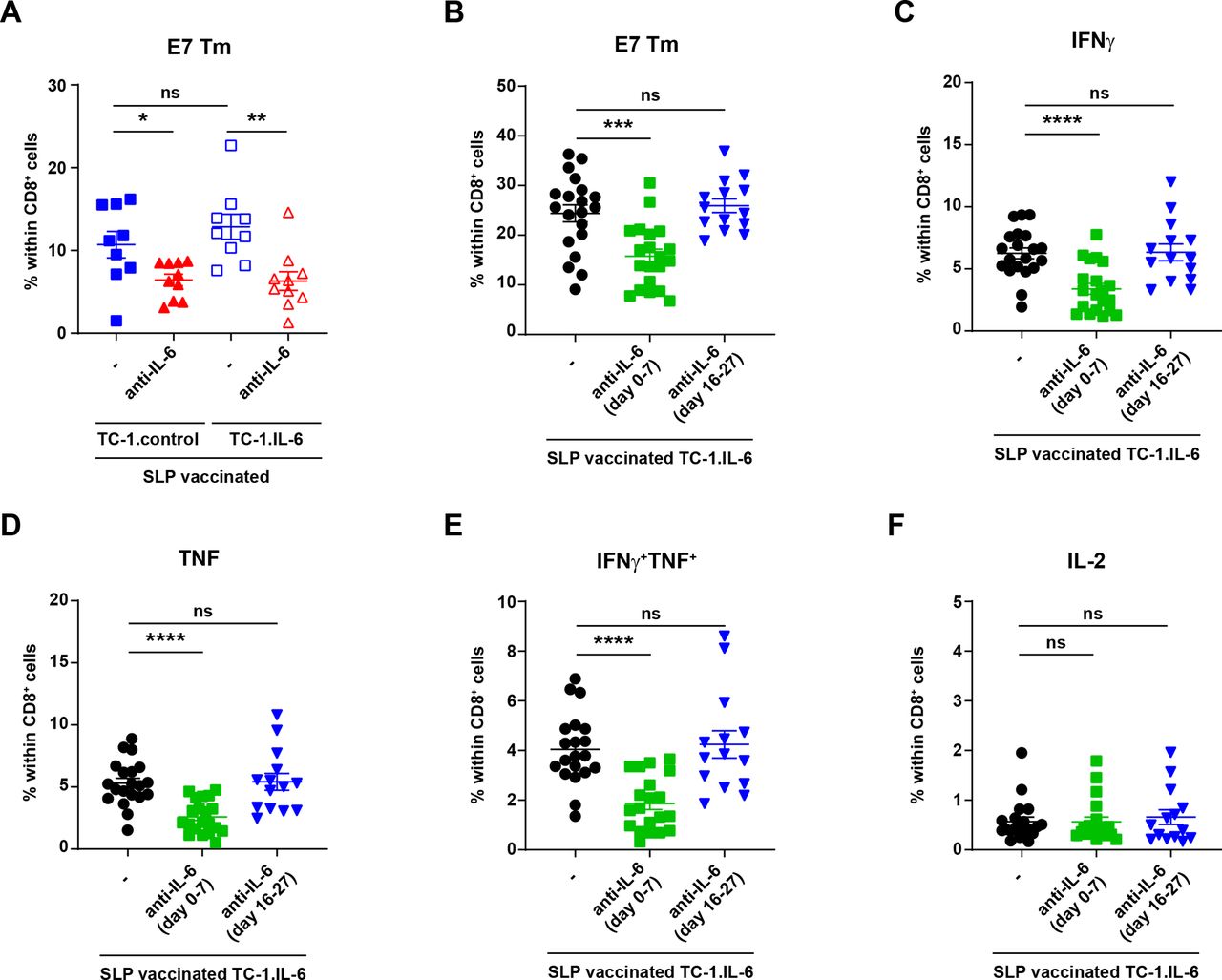
Thus, the IL-6 blockade-associated decrease in the magnitude of the vaccine-induced tumor-specific T cell response and in tumor-infiltration could neither be attributed to differences in the proliferative pool nor to an altered phenotype of CD4+ and CD8+ T cells during the regression phase. Therefore, we hypothesized that the vaccine-induced tumor-specific T-cell response was affected at the priming phase, either because IL-6R blockade interferes with IL-6 transpresentation by DC24 or because it blocked expansion of primed T cells.23 We used the antimouse IL-6 blocking antibody as this does not interfere with IL-6 transpresentation while it does block classical and trans-signaling of IL-6.24 IL-6 blockade efficiently blocked the IL-6 in serum (online supplemental figure S5) but also reduced the magnitude of the circulating HPV-specific CD8+ T-cell response in tumor-bearing mice measured at day 29 (figure 3A), indicating that a block of IL-6 transpresentation by DC was not the underlying cause for a lower HPV-specific T-cell response. Next, we examined if IL-6 blockade at the priming phase was the cause. TC-1.IL-6 tumor-bearing mice were injected with IL-6 blocking antibody before the priming phase (day 0–7) or after the tumor regression phase (day 16–27). Early but not late IL-6 blockade decreased the magnitude (figure 3B) and the production of IFNγ and/or TNF of the circulating tumor-specific T cell response measured at day 29 (figure 3C-F). These data indicate that blockade of IL-6 signaling during the time that CD8+ T-cell priming and tumor-infiltration occurs,14 has a strong effect on their expansion and effector function.

Early IL-6 blockade hampers the vaccine-induced T cell response in mice with IL-6 producing tumors. (A) The percentage of E7 HPV Tm+ cells within CD8+ cells in blood 29 days post-tumor challenge in SLP vaccinated TC-1.control and TC-1.IL-6 tumor-bearing mice with or without IL-6 blockade (day 8–29). (B–F) Mice were injected with TC-1.IL-6 tumor. Mice received anti-IL-6 from day 0 till 7 (day 0–7) or from day 16 to 27 (day 16–27) or none at all (-). All mice received the SLP prime and boost vaccine on day 8 and 22 post-tumor challenge, respectively. The percentage of E7 HPV Tm+ cells (B), IFNγ+ (C), TNF+ (D), IFNγ+ TNF+ (E) or IL-2 (F) producing CD8+ cells in blood at day 29 post-tumor challenge. Data are pooled from three independent experiments, yielding similar results. Graphs indicate mean values with SEM each dot represents data from an individual mouse. Significance between the two different groups in each tumor model (A) or between non-blocked versus blockade at one specific point (B–F) was determined by unpaired Student’s t-test. *P<0.05; **p<0.01; ***p<0.001; ****p<0.0001. IFNγ, interferon γ; IL-6, interleukin 6; ns, not significant; SLP, synthetic long peptide.
IL-6 signaling in intratumoral macrophages is required for full vaccine-induced tumor regression
Therapeutic vaccination-induced tumor infiltration by HPV-specific CD8+ T cells mediates the enhanced attraction of macrophages.14 To distinguish between indirect effects (as a consequence of a lower vaccine-induced T cell response) and direct effects of IL-6 blockade on myeloid cells, we made use of mice lacking the IL-6R on monocytes, macrophages and granulocytes, as IL-6 signaling is retained in T cells. For this reason, Il6rafl/fl×LysMcre+ transgenic mice and control Il6rafl/fl×LysMcre- mice (expressing IL-6R)25 were challenged with TC-1.control or TC-1.IL-6 tumor cells, vaccinated 8 days later and then followed in time. TC-1.control and TC-1.IL-6 tumors grew similarly in untreated mice, indicating that there are no intrinsic difference in tumor growth between Il6rafl/fl×LysMcre+, Il6rafl/fl×LysMcre- and wildtype mice (figure 4A). Following peptide vaccination, most of the TC-1.control and TC-1.IL-6 tumors regressed fully in the IL-6R proficient Il6rafl/fl×LysMcre- and wild-type mice. The tumors in mice lacking IL-6R expressing by these myeloid subsets showed fewer deep regressions after vaccination (figure 4B), an observation that was most prominent in TC-1.IL-6 tumor-bearing mice (figure 4C) and reflected in the lower percentage of complete regressions in TC-1.IL-6 tumor-bearing mice lacking IL-6R on their myeloid cell subset (figure 4D). This was not due to an altered infiltration with myeloid cells (online supplemental figure S6) or due to the granulocytic (Ly6G+) population within this myeloid subset as their depletion did not affect the efficacy of therapeutic vaccination in TC-1 bearing mice (online supplemental figure S7A-C) in contrast to macrophage depletion,14 26 indicating that this effect is due to the absence of IL-6R in macrophages. Importantly, the abrogation of complete regressions found in the Il6rafl/fl×LysMcre+ mice challenged with TC-1.IL6 is similar to what was found with respect to complete regression when IL-6 signaling was blocked by antibodies (figure 1E), suggesting that the lack of IL-6 signaling in macrophages but not that in the vaccine-induced T-cell response was the mechanism underlying the negative effect of IL-6 blockade on tumor regression. Indeed, there was no overt difference in the percentage or phenotype of the vaccine-induced tumor-specific CD8+ T-cell population in the circulation (online supplemental figure S8A–D) or among the total population of tumor-infiltrating tumor-specific CD8+ T cells (figure 4E). Intratumorally, no difference was observed in the phenotype of the infiltrating tumor-specific CD8+ T cells or the percentage of IFNγ producing CD8+ T cells (online supplemental figure S9A,B). In addition, the percentage or polarization of tumor infiltrating cytokine-producing CD4+ T cells or Tregs remained unchanged after vaccination between Il6rafl/fl×LysMcre+ and Il6rafl/fl×LysMcre- mice (online supplemental figure S9B,C). These data indicate that IL-6 signaling in macrophages is crucial for their role in the vaccine-induced full regression of tumors and prevention of recurrences.

IL-6 signaling in intratumoral macrophages is required for full vaccine-induced tumor regression. Tumor outgrowth graphs also indicating the number of alive mice from the total tested above the x-axis (A, B), the average tumor outgrowth (C; mean values with SEM), and the response rate according to RECIST criteria (D; NR, no response; PR, partial response; CR, complete response) of Il6rafl/fl×LysMcre+, Il6rafl/fl×LysMcre- and wild-type mice treated with SLP vaccine or kept untreated. Data are pooled from three independent experiments. (E) The percentage of intratumoral CD8+ cells within live gate (left) and the percentage of E7 Tm+ cells within CD8+ cells of Il6rafl/fl×LysMcre+, Il6rafl/fl×LysMcre- SLP vaccinated mice. Graphs indicate mean values with SEM each dot represents data from an individual mouse. Data are representative of one experiment. Significance was determined by unpaired Student’s t-test for difference in endpoint tumor size (day 22) in (C) and Mann-Whitney U test in (E), within each tumor model. *P<0.05; **p<0.01. IL-6, interleukin 6; SLP, synthetic long peptide.
IL-6R blockade downregulates SOCS3 and increases SIRPα on tumor-associated macrophages
The results obtained with the IL-6R deficient Il6rafl/fl×LysMcre+ mice unequivocally pinpointed altered IL-6 signaling in macrophages as the underlying cause for their impaired capacity to mediate tumor regression when IL-6 signaling was blocked by antibodies. An in-depth analysis of the myeloid cell composition in both tumor models with or without IL-6R blockade at the time of therapy-induced tumor regression revealed no difference in the percentage of CD11b+ myeloid cells (figure 5A, online supplemental figure S10A). Similarly, no differences were observed in the percentage of cDC2 CD11c+ and CD11c- immature (imm) macrophages. However, a small increase in monocyte-derived DCs (moDCs) was detected after IL-6R blockade in IL-6 producing tumors (figure 5B, online supplemental figure S10A). Subtle changes were observed in the percentage macrophages (F4/80+CD11b+) including iNOS+, Egr2+ and Ly6C+ macrophages (figure 5C, online supplemental figure S10B). In addition, the percentage of neutrophils (Ly6G+ CD11b+) remained unchanged (online supplemental figure S10C). The majority of macrophages expressed CD86, CD40 and MHC class II and although the percentage of macrophages expressing these markers is high already, IL-6 blockade increases this for CD40 and MHC class II in TC-1.IL-6 tumors (figure 5D). Finally, a small decrease in plasmacytoid DCs and small increase in CD103+ cDC1 cells was observed (figure 5E).

The composition of intratumoral myeloid cells is grossly unchanged with IL-6 axis blockade. The percentage of intratumoral CD11b+ myeloid cells (A), cDC2, moDCs, immature monocytes and monocytes (B), macrophages (C) and pDCs and cDC1 (E) in SLP vaccinated or untreated TC-1control and TC-1.IL-6 tumor-bearing mice with and without IL-6 blockade. Graphs indicate mean values with SEM each dot represents data from an individual mouse. Data are representative of one experiment. (D) The expression of CD86, CD40 and MHC class II on macrophages. Significance in differences in the percentage of the indicated vaccine-associated cell infiltration due to IL-6R blockade were determined by Student’s t-test within each tumor model (A–E). *P<0.05; **p<0.01; ***p<0.001. IL-6, interleukin 6; mo DCs, monocyte-derived dendritic cells; ns, not significant; pDC, plasmacytoid DCs.
As no overt alterations were observed in myeloid cell frequencies that could explain the negative effect of IL-6 blockade, we analyzed the phenotype of these subsets. SOCS3 expression in macrophages is required to suppress, among others, the anti-inflammatory properties of IL-6 while retaining its proinflammatory properties and is thus essential for effective M1-type activation and function in macrophages.27–30 IL-6R blockade decreased the percentage of macrophages expressing SOCS3 as well as the levels of SOCS3 but not that of SOCS1, on total CD11b+ cells and most prominently in TC-1.IL-6 infiltrating macrophages (figure 6A and online supplemental figure S11A). SOCS3 but not SOCS1 downregulation was also observed in other myeloid cells infiltrating TC-1.IL-6 tumors (online supplemental figure S11B,C). The IL-6R blockade mediated SOCS3 downregulation coincided with prolonged STAT3 activation but not STAT1 signaling, evidenced by the higher expression of pSTAT3 TC-1.IL-6 tumor-infiltrating macrophages (figure 6B and C), a feature previously reported for SOCS3 knock-out macrophages after IL-6 stimulation,31 suggesting that the anti-inflammatory program of macrophages was activated when the IL-6R was blocked.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IL-6 blockade decreases the levels of SOCS3 and increases SIRPα levels. (A) The percentage of intratumoral SOCS3+CD11b+ (myeloid cells) and SOCS3+F4/80+ (macrophages) cells and mean fluorescent intensity (MFI) of SOCS3 on macrophages. (B) MFI of pSTAT3 and pSTAT1 on total CD11b+ myeloid cells and F4/80+CD11b+ macrophages. (C) MFI of SIRPα on F4/80+CD11b+ macrophages and CD11b-CD103+ (cDC1) cells. Scatter plots indicate mean values with SEM each dot represents data from an individual mouse. Violin plots show the full distribution of the data, each dot representing an individual mouse. Data are representative of one experiment. Significance (A–C) in differences in the indicated vaccine-associated percentage of indicated cell infiltration or mean fluorescence intensity of indicated markers on cells due to IL-6R blockade was determined by Student’s t-test within each tumor model. Significance between groups in D is determined by one-way ANOVA. *P<0.05; **p<0.01; ****p<0.0001. ANOVA, analysis of variance; SIRPα, signal-regulatory protein alpha; SOCS3, suppressor of cytokine signaling 3.
SIRPα is abundantly expressed by macrophages and negatively regulates phagocytosis of host cells by these macrophages as well as the production of proinflammatory cytokines.32 Therapeutic vaccination resulted in a decreased percentage of CD11b+ cells, including macrophages, expressing SIRPα in TC-1.control tumors (figure 6D and online supplemental figure S12), in line with a proinflammatory activation of macrophages.32 Tumor-produced IL-6-mediated activation of macrophages resulted in partial decrease of SIRPα expression by macrophages and this was not decreased to the levels observed in TC-1.control tumors when TC-1.IL-6 bearing mice were vaccinated (figure 6D and online supplemental figure S12). Importantly, IL-6R blockade increased SIRPα expression on macrophages, most prominently in TC-1.IL-6 tumors (figure 6D and online supplemental figure S12). IL-6 blockade did not change SIRPα expression on CD11b+ DC, Ly6Chi CD11b+ cells and monocytes but was increased on CD103+ DC, suggesting that local cross-presentation of tumor antigens might be lowered (figure 6D and online supplemental figure S12). Decreased levels of SOCS3 and increased levels of SIRPα after IL-6 blockade have strong impact on the tumoricidal function of macrophages31 32 and are likely to explain why IL-6R blockade is not able to improve the efficacy of therapeutic vaccination in this model.
Discussion
In this study, we applied IL-6 and IL-6R blockade in an IL-6-mediated immunotherapy-resistant TC-1 tumor model.10 Unexpectedly this did not resolve therapy resistance, instead it exacerbated tumor outgrowth, reflected by fewer complete regressions and more recurrences. Blockade of IL-6 signaling had a direct effect on the expansion and migration of tumor-specific CD8+ T cells forming one explanation for a lack of full tumor regression. However, the failure of therapeutic vaccination to induce full tumor regression in mice lacking the IL-6R on macrophages despite the strong presence of CD8+ effector T cells pointed out that the main underlying mechanism was the malfunction of macrophages at the time of therapeutic vaccination-induced tumor regression. Most likely this was driven by a switch from an IL-6 induced proinflammatory program towards an anti-inflammatory program a result of an IL-6R blockade-mediated decreased expression of SOCS3.27–30 In addition, IL-6R blockade instigated the upregulation of SIRPα, which is known to interfere with the macrophage-mediated phagocytic clearance of malignant cells.33 Therapy failure due to an inadequate intratumoral macrophage response, despite the presence of fully activated intratumoral tumor-specific CD8+ T cells, fits with earlier reports demonstrating that macrophages are required for TC-1 tumor regression after therapeutic vaccination14 26 and functions as a secondary immune escape mechanism.34 Notably, while these effects were most prominently demonstrated in the context of high levels of tumor-produced IL-6, similar observations were made in the TC-1 control tumor model, which produced much lower levels of IL-6.
We previously showed that IL-6 production by tumors altered the function of spontaneously tumor-infiltrating DCs and macrophages, reflected by a lower expression of MHC class II by these myeloid cell subsets.10 Therapeutic vaccination induced tumor infiltration by IFNγ-producing T cells, partly restored the expression of MHC class II by macrophages in TC-1.IL-6 tumors and stimulated their initial tumor regression but failed to cure mice as all tumors recurred.10 Our current data do not oppose our earlier findings but rather fine tunes the chain of events. Clearly, the overexpression of IL-6 had less of an effect on the initial T cells and macrophage-driven tumor regression,10 fitting with IL-6’s positive role shown here, but it did foster tumor recurrence through a myriad of mechanisms we do not understand yet.
IL-6 participates in two modes of receptor signaling, the context in which it is blocked thus may affect outcome.35 This is illustrated by several studies demonstrating that antibody-mediated blockade of IL-6 signaling during tumor outgrowth, generally in combination with PDL1 blockade, increased the spontaneously induced tumor-reactive T cell response with as result an immune-mediated tumor growth retardation in several murine tumor models.7 11 12 Also in a setting where therapeutic vaccination, in combination with checkpoint blockade and TGFβ blockade delayed TC-1 tumor growth, a positive effect of co-treatment with IL-6 blocking antibodies was observed.13 In our model, therapeutic vaccination of TC-1.IL-6 bearing mice resulted in a strong and fast tumor regression and cure of about 40% of mice. IL-6R blockade reduced this percentage to about 0%–10% when different IL-6R blockade schedules were applied. Whereas in the other models IL-6 blockade is applied in a setting of a chronic inflammatory process where tolerance rules the roost, therapy-driven tumor regression is a process of tissue destruction associated with acute inflammation, also found in allograft rejection, pathogen clearance and flares of autoimmunity.15 Indeed, IL-6 was found to drive the development of type 1 T cell responses, the migration of T cells and monocytes to the site of inflammation, and the activation of the tumoricidal function of macrophages and neutrophils in settings of acute peritoneal inflammation, acute joint inflammation, acute colitis and fungal infection,21–23 36 37 sustaining our observations with respect to the role of IL-6 in therapeutic vaccine-driven T-cell expansion and macrophage activation. The cascade that leads to tissue destruction is the recruitment of antigen-specific T cells to the site of acute inflammation, these T cells then secrete ligands to recruit innate immune effector cells. At this point several cytotoxic mechanisms converge on the target tissue, and its complete destruction occurs through the activated effects of adaptive and innate killer cells, including cytotoxic T cells (CTLs) and macrophages.15 Indeed our previous studies in the TC-1 tumor model revealed that the vaccine-induced T cells recruited tumoricidal macrophages to the tumor and not vice versa.14 The lack of a difference in the percentages of tumor-infiltrating CD8 +T cells and macrophages when TC-1.control is compared with TC-1.IL-6 and the slight decrease in the number of tumor-infiltrating tumor-specific CD8 T cells but not that of macrophages, supports the notion that the vaccine-induced migration of macrophages is not altered when the levels of IL-6 are increased. Furthermore, the unaltered intratumoral tumor-specific CD8 T cell response in mice with IL-6R-deficient macrophages argues against a role for IL-6 signaling in macrophages to stimulate T cell antitumor activity, and the lack thereof being the cause for suboptimal tumor regression. Most likely, IL-6 signaling in these macrophages is required for them to exert a direct cytotoxic effect on tumor cells. This is supported by our data showing less complete tumor regressions despite the presence of effector CD8 T cells in vaccinated mice lacking the IL-6R on macrophages as well as by a study showing that IL-6 drives both pathogen-specific type 1 T cell responses and activates the cytotoxic activity in macrophages when SOCS3 expression in these cells increased.38 Indeed, we observed an IL-6 mediated increased expression of SOCS3 in the intratumoral macrophages, suggesting that it armed them with cytotoxic potential.
Tumor-infiltrating tumor-reactive T cells function as a tissue-specific trigger for tumor destruction but are not always necessary39 or sufficient15 for tissue damage. We and others have shown that the T-cell-driven regression of tumors may require the attraction and activation of tumoricidal phagocytic macrophages14 26 34 or neutrophils40 to deliver the final cytotoxic insult. While neutrophils display antitumor activity in the TC-1 model,40 we showed here that they do not contribute to the efficacy of therapeutic vaccination. The requirement of T cells to deliver the trigger and macrophages to fully execute the cytotoxic program in cancer cells fits well with the concept of programmed cell removal, linking programmed cell death to the removal of the dying cell.41 The importance of this mechanism is illustrated by the identification of several phagocytosis checkpoints, the expression of which foster tumor immune escape.33 In our model, IL-6R blockade resulted in the increased expression of SIRPα by macrophages, suggesting that this checkpoint may have played a role in the negative effects of IL-6R blockade. We did not measure the expression of other phagocytosis-regulating checkpoints expressed by macrophages, such as PD-1 and LILRB,33 42 but do not exclude that they play a role too.
Conclusions
Blockade of the IL-6 signaling pathway has provided modest benefit in the immunotherapy of cancer. Our data suggest that IL-6 signaling is critical for macrophage function under circumstances of immunotherapy-induced tumor tissue destruction, in line with the acute inflammatory functions of IL-6 signaling described in infections. This suggests that when the context changes in which IL-6 blockade is given, for instance during successful PD-1:PD-L1 blocking-mediated tumor regressions, it may result in suppression of IL-6-driven proinflammatory actions. New studies should focus on how to maintain SOCS3 expression in macrophages and blockade of phagocytic checkpoints such as SIRPα during IL-6 signaling blockade.
Data availability statement
Data are available on reasonable request. The TC-1.control and TC-1.IL-6 cell lines are available on request.
Ethics statements
Ethics approval
All animal experiments were approved by the Animal Experiments Committee of LUMC and were executed according to the animal experimentation guidelines of LUMC in compliance with the guidelines of Dutch and European committees.
Acknowledgments
We like to thank Stefan Rose-John (Kiel University, Germany) and Christoph Garbers (Otto-Von-Guericke-University Magdeburg, Germany) for their insights and input during the initiation of our study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors SHvdB, TvH, RA, EBN, MvE and JWK designed the project and experiments and interpreted the results. EBN, MvE, CL and JWK performed experiments. EBN and KLMCF designed and produced the tumor cell lines. H-WM, SH, TK provided expert feedback on IL-6, IL-6 blockade and provided the Il6rafl/fl×LysMcre+ and Il6rafl/fl×LysMcre- mice. EBN, SHvdB, RA, TvH wrote the manuscript. All authors read and approved the final manuscript.
Funding This work was supported by a grant from the Dutch Cancer Society (KWF 2015-7824) and the Oncode Institute Base Fund to SHvdB.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.