Article Text

Abstract
Background Emerging data suggest predictive biomarkers based on the spatial arrangement of cells or coexpression patterns in tissue sections will play an important role in precision immuno-oncology. Multiplexed immunofluorescence (mIF) is ideally suited to such assessments. Standardization and validation of an end-to-end workflow that supports multisite trials and clinical laboratory processes are vital. Six institutions collaborated to: (1) optimize an automated six-plex assay focused on the PD-1/PD-L1 axis, (2) assess intersite and intrasite reproducibility of staining using a locked down image analysis algorithm to measure tumor cell and immune cell (IC) subset densities, %PD-L1 expression on tumor cells (TCs) and ICs, and PD-1/PD-L1 proximity assessments.
Methods A six-plex mIF panel (PD-L1, PD-1, CD8, CD68, FOXP3, and CK) was rigorously optimized as determined by quantitative equivalence to immunohistochemistry (IHC) chromogenic assays. Serial sections from tonsil and breast carcinoma and non-small cell lung cancer (NSCLC) tissue microarrays (TMAs), TSA-Opal fluorescent detection reagents, and antibodies were distributed to the six sites equipped with a Leica Bond Rx autostainer and a Vectra Polaris multispectral imaging platform. Tissue sections were stained and imaged at each site and delivered to a single site for analysis. Intersite and intrasite reproducibility were assessed by linear fits to plots of cell densities, including %PDL1 expression by TCs and ICs in the breast and NSCLC TMAs.
Results Comparison of the percent positive cells for each marker between mIF and IHC revealed that enhanced amplification in the mIF assay was required to detect low-level expression of PD-1, PD-L1, FoxP3 and CD68. Following optimization, an average equivalence of 90% was achieved between mIF and IHC across all six assay markers. Intersite and intrasite cell density assessments showed an average concordance of R2=0.75 (slope=0.92) and R2=0.88 (slope=0.93) for breast carcinoma, respectively, and an average concordance of R2=0.72 (slope=0.86) and R2=0.81 (slope=0.68) for NSCLC. Intersite concordance for %PD-L1+ICs had an average R2 value of 0.88 and slope of 0.92. Assessments of PD-1/PD-L1 proximity also showed strong concordance (R2=0.82; slope=0.75).
Conclusions Assay optimization yielded highly sensitive, reproducible mIF characterization of the PD-1/PD-L1 axis across multiple sites. High concordance was observed across sites for measures of density of specific IC subsets, measures of coexpression and proximity with single-cell resolution.
- programmed cell death 1 receptor
- breast neoplasms
- lung neoplasms
- immunohistochemistry
- biomarkers
- tumor
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
PD-1/PD-L1 immune checkpoint inhibition has revolutionized cancer treatment. However, the majority of patients unfortunately still do not respond. There is a need for predictive assays that can be used to determine which therapeutic regimen is most likely to benefit a given patient. The most commonly used approach for preselecting patients for anti-PD-(L)1 therapy is single-stain chromogenic immunohistochemistry (IHC) for PD-L1 expression. There are now numerous FDA-approved assays that test for PD-L1 expression within the pretreatment tumor microenvironment (TME).1
PD-L1 IHC assays enrich for response to PD-1/L1 blockade; however, PD-L1 IHC is imperfect. Approximately 10%–15% of patients with PD-L1-negative tumors may respond to therapy, and ~50% patients with PD-L1+ tumors do not respond.2 There are also other challenges associated with the current PD-L1 testing environment. The numerous PD-L1 IHC assays in use employ different scoring algorithms. Some score membranous PD-L1 expression on tumor cells (TCs) only, some focus on immune cell (IC) PD-L1 expression, while yet others assess a combination of these features.3 Notably, pathologists have poor interobserver concordance when attempting to score PD-L1 expression on ICs, especially in low expression ranges.4 PD-L1 can also be expressed in the TME by both adaptive and constitutive mechanisms,5 and it is thought that anti-PD-1/PD-L1 acts primarily on those cases with an adaptive mechanism of display.6 Such an adaptive pattern of PD-L1 expression is typically represented in the TME by detecting PD-1 adjacent to PD-L1, and accordingly, biomarkers representing their combined expression in close proximity show improved predictive ability compared with those that measure PD-L1 expression alone.7 8
Multispectral, multiplex immunofluorescent (mIF) imaging approaches are capable of characterizing the TME in a way that overcomes the limitations detailed above. Multispectral mIF allows for the simultaneous quantitative characterization of six to eight markers across a single formalin-fixed paraffin-embedded tissue section. Application of this technology to characterizing PD-1/PD-L1 axis expression can thus aid in the accurate quantification of %PD-L1 expression across the TME as well as identify whether it is a TC or IC expressing PD-L1. It also allows for characterization of the ‘spatial biology’ of a tumor sample, such as interrogating PD-1/PD-L1 cell-to-cell spatial interactions within the TME. Initial studies from individual institutions on tumor specimens from patients with non-small cell lung cancer (NSCLC),9 10 head and neck squamous cell carcinoma,11 Hodgkin lymphoma,12 Merkel cell carcinoma,8 13 and melanoma,7 14 15 among others, reinforce the potential of mIF to detect spatially resolved immunoactive features within the TME and associating these findings with clinical outcomes.
Before mIF technology could potentially be translated into clinical practice, it is vital to standardize and validate an end-to-end workflow that supports multisite trials and clinical laboratory processes. To that end, an optimized six-plex mIF assay for characterizing the PD-1/PD-L1 axis was developed. The assay included markers for PD-1, PD-L1, CD8, FoxP3, cytokeratin (CK) (tumor marker), and CD68 and was optimized using rigorous, quantitative assessments of equivalence to chromogenic IHC staining, that is, the current clinical ‘gold standard’.16 A total of six laboratories participated, including Johns Hopkins University, Yale University, MD Anderson Cancer Center, Earle A. Chiles Research Institute, Akoya Biosciences, and Bristol-Myers Squibb. Reproducibility was assessed within and across sites using control tissues and tissue microarrays (TMAs) of breast carcinoma and NSCLC. Concordance was tested for measurements of cell densities, %PD-L1 coexpression by cell type (TC vs IC), and PD-1/PD-L1 proximity.
Methods
Study design
Six laboratories participated in the development and assessment of intersite and intrasite staining reproducibility and six-plex mIF assay concordance for quantifying the PD-1/PD-L1 axis. Each site was supplied with the same assay reagents, and serial sections from tonsil tissue and TMAs for breast cancer and NSCLC were distributed to each site. Each laboratory stained their allotment of slides in two different staining batches to facilitate assessments of intrasite as well as intersite reproducibility. Slides were imaged at each site in order to qualitatively confirm successful staining. Stained slides were then shipped to a single site for final multispectral image acquisition and subsequent quantitative data analysis. The image analysis was conducted in a blinded fashion to avoid potential bias related to study site.
Pathology specimens
Formalin-fixed paraffin-embedded tissue (FFPE) sections from archival tonsil tissue and the breast and NSCLC TMAs were cut in 4 µm serial sections onto positively charged slides. The NSCLC TMA block consisted of 144 cores, and the breast cancer TMA block contained 168 cores. Each core was 0.6 mm in diameter and represented an individual patient. Three of the cores on each of the two TMAs were used as on-slide controls for setting thresholds of PD-L1 positivity. TMAs were supplied by Yale Pathology Tissue Services (New Haven, Connecticut, USA). Each of the six study sites received 10 tonsil slides, two slides from the breast cancer TMAs, and two slides from the NSCLC TMAs. For a detailed description of tissue section serialization and distribution, please see online supplemental table 1.
Supplemental material
mIF assay reagents
Primary antibodies included those to CD8, CD68, FoxP3, pan-CK (clone AE1/3), PD-1, and PD-L1 (table 1). All sites used primary antibodies from the same lot. For CD8 and CK: Akoya’s Opal Polymer anti-mouse and -rabbit HRP (1:5, ARH1001EA) was used for secondary detection. Leica Biosystems PowerVision Poly-HRP antimouse was used for FoxP3 and CD68 (50%, PV6114, Leica Biosystems) and Poly-HRP anti-rabbit was used for PD-1 and PD-L1 (50%, PV6119, Leica). Each site received an Opal 7-color Automated IHC Detection Kit (NEL821001KT, Akoya Biosciences, Marlborough, Massachusetts, USA) containing the following TSA fluorophores: Opal 520, Opal 540, Opal 570, Opal 620, Opal 650, Opal 690, and spectral DAPI. All fluorophores and DAPI were prepared according to manufacturer guidelines.
Final, optimized six-plex mIF assay conditions for characterizing the PD-1/PD-L1 axis
mIF Assay Development and Staining
The six-plex mIF assay was optimized as previously described.15 In brief, for each antibody, staining parameters were first optimized using single stain, chromogenic IHC on tonsil sections. Next, each primary antibody was paired to a select TSA fluorophore and single stain, that is, ‘monoplex’ IF staining was performed. TSA fluor-marker pairings were based on known brightness rankings, with more abundant markers paired with less bright fluorophores (Opals 570, 620, and 690). TSA dilutions started at 1:150 and were titrated to achieve the recommended target range of 10–30 in normalized brightness counts, provided that a sensitivity equivalent to chromogenic IHC was maintained. Ten multispectral 20× high power fields (HPFs) were then acquired from five archival NSCLC specimens (total of 50 HPFs) using the Vectra Polaris. The HPFs were carefully aligned across serial sections for equivalence assessments of IF to IHC to ensure measurements were of the same tissue morphological regions. Equivalency was based on image analysis-based counts of cells positively stained for each of the six markers/total cells in each HPF, that is, % positive cells for each marker, using the inForm Tissue Finder cell phenotyping function. Of note, the cell counting algorithm for the chromogenic IHC images was different from the algorithm trained to count cells in the monoplex and multiplex IF because the imagery differs based on how it was acquired. For markers FoxP3, CD68, PD-1, and PD-L1, it was necessary to change the secondary detection system from Opal Polymer anti-mouse and -rabbit HRP to the Leica PowerVision Poly-HRP IHC Detection system to achieve equivalent sensitivity to chromogenic IHC.
Following the successful conversion of the chromogenic protocols to immunofluorescence, all the monoplex immunofluorescence protocols were combined to form a complete six-plex, seven-color assay panel. The standard seven-color TSA protocol template on the BOND RX was used with modifications. Modifications included that tissues underwent an initial antigen retrieval step of ER2 at 100°C for 40 min, a double dispensing of the TSA reagents (incubation time of 0 and 10 min), and that diamidino-2-phenylindole (DAPI) was double dispensed at a volume of 150 µL. Adjustments to the staining order were made based on quantitative assessment of equivalency to the monoplex imagery. The final protocol used to stain the tissues is provided in table 1.
mIF Staining, Multispectral Image Acquisition and Quantitative Analysis
All tonsil sections and TMAs underwent an initial 3-hour baking step at 65°C. During this initial baking step, slides were held in a slide rack in a vertical manner for the first 1.5 hours. They were then rotated to sit horizontally for the second 1.5 hours. A second bake and de-wax step was then performed using a dewax solution (AR9222, Leica Biosystems) on the BOND RX to ensure that all paraffin was removed.
Slides were then stained using the aforementioned optimized, automated mIF staining protocol. Multispectral images were acquired using the Vectra Polaris Automated Quantitative Pathology Imaging System. A set of library slides were created in order to achieve accurate spectral unmixing and data quantification of each Opal fluorophore in inForm. Specifically, a library was generated by staining serial sections of tonsil tissue with CD20 (clone L26, PM0044AA, Biocare Medical) and each individual fluorophore. Additionally, a tonsil serial section was stained with DAPI and added to the library. Such an approach facilitates the capture of pure emission spectra, which are then used in the unmixing process. Lastly, a section that did not have any stain applied was used to capture the background tissue autofluorescence. Prior to processing, all images were assessed for quality control. Criteria for rejection included poor tissue quality, such as tissue folds or missing tissue sections, and staining artifacts, including signal dropout and air bubbles.
For each project, all HPF images were processed and analyzed with inForm software (V.2.4.10). A single algorithm for spectral unmixing, cell segmentation, cell classification, that is, ‘phenotyping’, and quantification of expression intensity was developed for each tissue type (tonsil, breast and NSCLC), and the same algorithm was applied by a single site to all the cores within and across TMAs for each tumor type. As a part of this process, cells were segmented into cytoplasmic, nuclear, and membrane compartments. For the purposes of determining whether a cell was positive for a given marker, signal levels for CD8, PD-L1, and PD-1 were measured in the membrane compartment, while CD68 and CK were measured in the cytoplasmic compartment. Lastly, FoxP3 signal levels were measured in the nuclear compartment. Once all images were processed, the data were exported for further analysis of IC densities, PD-L1 expression by cell type, and PD-1/PD-L1 proximity in the R-script package phenoptrReports (Akoya BioSciences).
mIF staining reproducibility on tonsil serial sections
Following an overview scan, 12 matching 20× HPFs were selected on the 60 tonsil serial sections: four from the cortex, four from the crypt/mantel, and four from the follicle. These microanatomic regions were selected to capture areas enriched for the markers of interest, that is, cortex: CD8 and FoxP3; crypt: PD-L1 and CK; and the follicle: CD68 and PD-1. Cells phenotyped as ‘positive’ for each marker per HPF were aggregated, and the average of the top quartile of signal intensity was determined. This approach was chosen for its sensitivity in highlighting potential variability in staining performance.
Intersite and intrasite percent coefficients of variation (%CV) were determined for each marker. First, an average cell number/HPF for each marker was calculated for four HPFs on each slide. The average cell numbers per slide were then used to calculate intersite and intrasite %CVs. The intersite %CV for each marker was determined by first calculating the %CV of average cell numbers in six serial sections distributed across the six sites (one slide per site), for a total of five groups. The %CVs for each marker were then averaged across the five groups, and an intersite %CV was calculated for each marker (online supplemental figure S1A). Intrasite %CV for each marker was determined by first calculating the %CV for average cell number per HPF across five serial sections from each site. The %CVs from each site were then averaged (online supplemental figure S1B).
Intersite and intrasite concordance for cell density assessments using TMAs
Densities (number of cells expressing a given marker/tissue area (mm2)) of PD-L1, PD-1, CD68, CD8, CK, and FoxP3 cells in each core from the breast and NSCLC TMAs were determined for each batch for each site. Intersite concordance assessments were determined by averaging the cell densities for run 1 and run 2 for each TMA core for each site. Averaged TMA core cell densities were then plotted against their respective counterparts for every site. Linear regression analysis was run, and the slope, intercept, and R2 values were calculated. Any TMA core data that did not have an accompanying counterpart was excluded from the analysis. The total intersite R2 value and slope concordance for each marker were calculated by averaging all R2 values and slopes from each site-to-site comparison.
Intrasite concordance compared the same TMA cores for each site using run 1 data points as X and run 2 data points as Y. A simple linear regression was plotted onto the data to determine the slope, intercept, and R2 value. Any cores that did not have both run 1 and run 2 data were removed from subsequent analysis. The total intrasite R2 value and slope were determined by averaging across all sites for each marker.
Intersite concordance of percent PD-L1 expression and PD-1/PD-L1 proximity analysis
The number of cells displaying the following markers and marker combinations were determined for each TMA core: PD-L1+ cells, PD-1+ cells, CD68+ cells, CK+ cells, CD68+/PD-L1+ cells, and CK+/PD-L1+ cells. For the combinations, a threshold was applied to the measured PD-L1 signal in each CK+ phenotype and each CD68+ phenotype to assign a cell as PD-L1+ versus PD-L1−. Three cores on each slide of the breast and the lung TMAs were selected to serve as on-slide controls. The threshold was normalized to the on-slide tissue controls to adjust for potential batch-to-batch variation across sites and set thresholds of positivity. Percent PD-L1 positivity was calculated by the following calculation for each TMA core [(colocalized phenotype/single phenotype) * 100]. Site-to-site percentages for %PD-L1 expression by CK+ TCs and CD68+ macrophages were graphed, and using simple linear regression, the R2 value and slope were interpolated. The total R2 and slope for %PD-L1/CD68+ and %PD-L1/CK+ were calculated by averaging all intersite values.
The number of PD-1 cells within a 25 µm radius of a PD-L1 cell was determined for every TMA core from each site using phenoptrReports. Intersite concordance agreement was evaluated by determining the slope and fit (R2) of a linear regression to scatter plots of data. The average fit and slope were calculated by averaging all intersite values.
Statistical analysis
All data were analyzed and graphed using both Excel and GraphPad Prism (V.8.3.0, GraphPad Company, San Diego, California, USA). Data analysis was performed using R software V.3.6.3 with built-in packages and custom routines. P values <0.05 were considered statistically significant.
Results
Multiplex fluorescence assay staining and validation against conventional chromogenic IHC
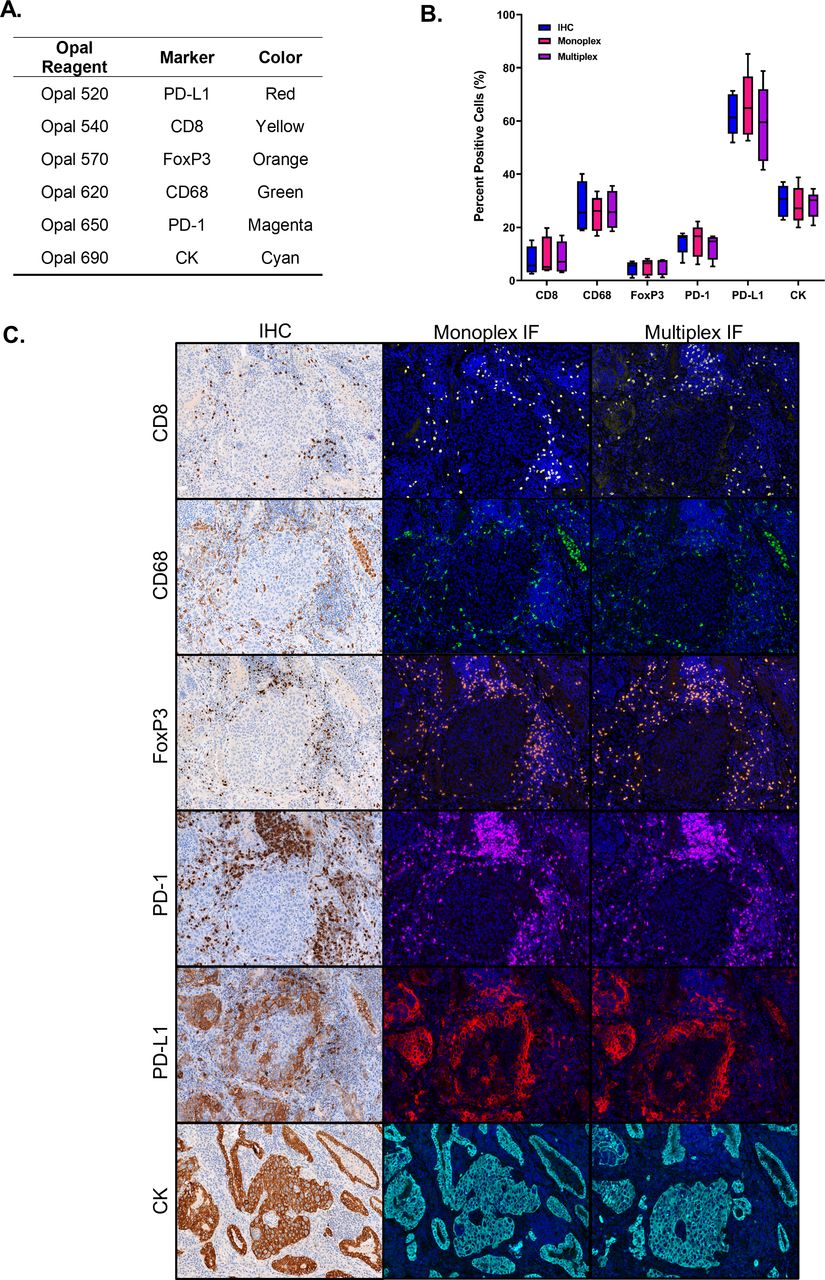
The objective of this step was to optimize a multispectral mIF panel to achieve equivalent sensitivity to chromogenic IHC for each individual marker. Markers were paired with Opal fluorophores that complimented their abundance and spatial location (figure 1A), and monoplex IF stains were tested for equivalence to chromogenic IHC. Four of the six markers (CD68, FoxP3, PD-1, and PD-L1) required the use of Leica’s Powervision HRP secondary to achieve the same sensitivity as the optimized chromogenic IHC. The markers were then combined into the multiplex format, and the percent positive cells for each marker between chromogenic DAB, monoplex IF, and mIF demonstrated equivalence across all three staining modalities (figure 1B and C). The assay took approximately 3–4 months to optimize by the lead site. After it was optimized, the protocol was provided to the other five laboratories, where it was used without additional modification.

The multiplex immunofluorescent (mIF) assay is comparable with monoplex IF and ‘gold standard’ chromogenic IHC staining. (A) Six-plex mIF assay reagents including the TSA-Opal and marker pairings, as well as the clone used for detecting each target. (B) Quantitative comparison of percentage of cells phenotyped as ‘positive’ for each marker by staining approach (chromogenic IHC, monoplex IF, and multiplex IF). For each marker, 10 HPFs per sample (n=5 NSCLC archival specimens) were acquired, and the % positive cells were averaged. Plot shows median and IQR, with whiskers showing min to max for each marker. (C) Representative images for each marker showing comparable staining patterns and cell densities on sequential NSCLC slides stained with chromogenic IHC stains, monoplex IF, and the mIF assay. HPFs, high power fields; IF, immunofluorescent; IHC, immunohistochemistry; NSCLC, non-small cell lung cancer.
Intersite and intrasite reproducibility of mIF assay in tonsil sections
Serial sections of tonsil stained with mIF by each of the six sites were evaluated for expression of each marker in the assay (figure 2A and B). The average intersite staining coefficient of variation (CV) across all sites was 20% for the top quartile of expression intensity, with CD8 and FoxP3 displaying higher %CVs compared with the other markers (figure 2C). Staining assessment revealed an average total intrasite %CV of 10% across all six markers, with a maximum CV of 13% (figure 2C), indicating minimal variability of staining within each site.

Intersite and intrasite reproducibility for the six-plex mIF assay in tonsil tissue. (A) Representative low power images from tonsil serial sections stained at each site.* Yellow=CD8, orange=FoxP3, green=CD68, magenta=PD-1, red=PD-L1 and cyan=CK (tumor marker). (B) High power photomicrographs corresponding to white boxes in low-power images showing staining patterns in the tonsillar crypts (left) and follicles (right). (C) Average intersite and intrasite CVs for each marker, as well as an average %CV for all markers. These comparisons were performed on only the top quartile of cells for each marker to provide a sensitive measure of potential variability. *Site 5 was excluded from this comparison due to a combination of mIF assay run failure and delayed data submission. mIF, multiplex immunofluorescent.
Intersite and intrasite concordance for assessments of cell densities in tumor TMA sections
Once intersite and intrasite agreement was achieved on tonsil, two serial sections of breast cancer TMAs and lung cancer TMAs were stained at each of the six sites in two separate batches (run 1 and run 2). Strong concordance in mIF staining patterns in tumor tissues was observed across all sites and batches (figure 3A). Intersite concordance plots for cell densities of PD-L1, PD-1, CD68, CD8, FoxP3, and CK were generated and consistent agreement was observed across all sites for each marker and in both tumor types (figure 3B and C, online supplemental figures S2 and S3). The one exception was intersite and intrasite reproducibilities for CD68 in NSCLC, which showed an average R2 value of 0.47 and a slope of 0.54 and R2 of 0.67 and slope of 0.60, respectively. This is most likely due to the challenges of segmenting and subsequent enumeration of the CD68+ macrophages, which often display irregular cell shapes. The intrasite concordances were slightly higher than the intersite concordances. For example, the average intrasite agreement on the breast TMA among CD68 and FoxP3 was R2=0.83 (slopes=0.90 and 0.89), with PD-L1, PD-1, CK and CD8 having R2 values of 0.85 (slope=0.88), 0.93 (slope=0.87), 0.93 (slope=0.93) and 0.94 (slope=1.01), respectively (figure 3C). The average intersite concordance for PD-L1, PD-1, CD68, CD8, and FoxP3 had R2 values ranging from 0.67 to 0.89 (slopes of 0.89–1.10), with PD-1 displaying the strongest fit. The NSCLC TMA core imagery, intrasite, and intersite concordance cell density data are provided in online supplemental figures S3.
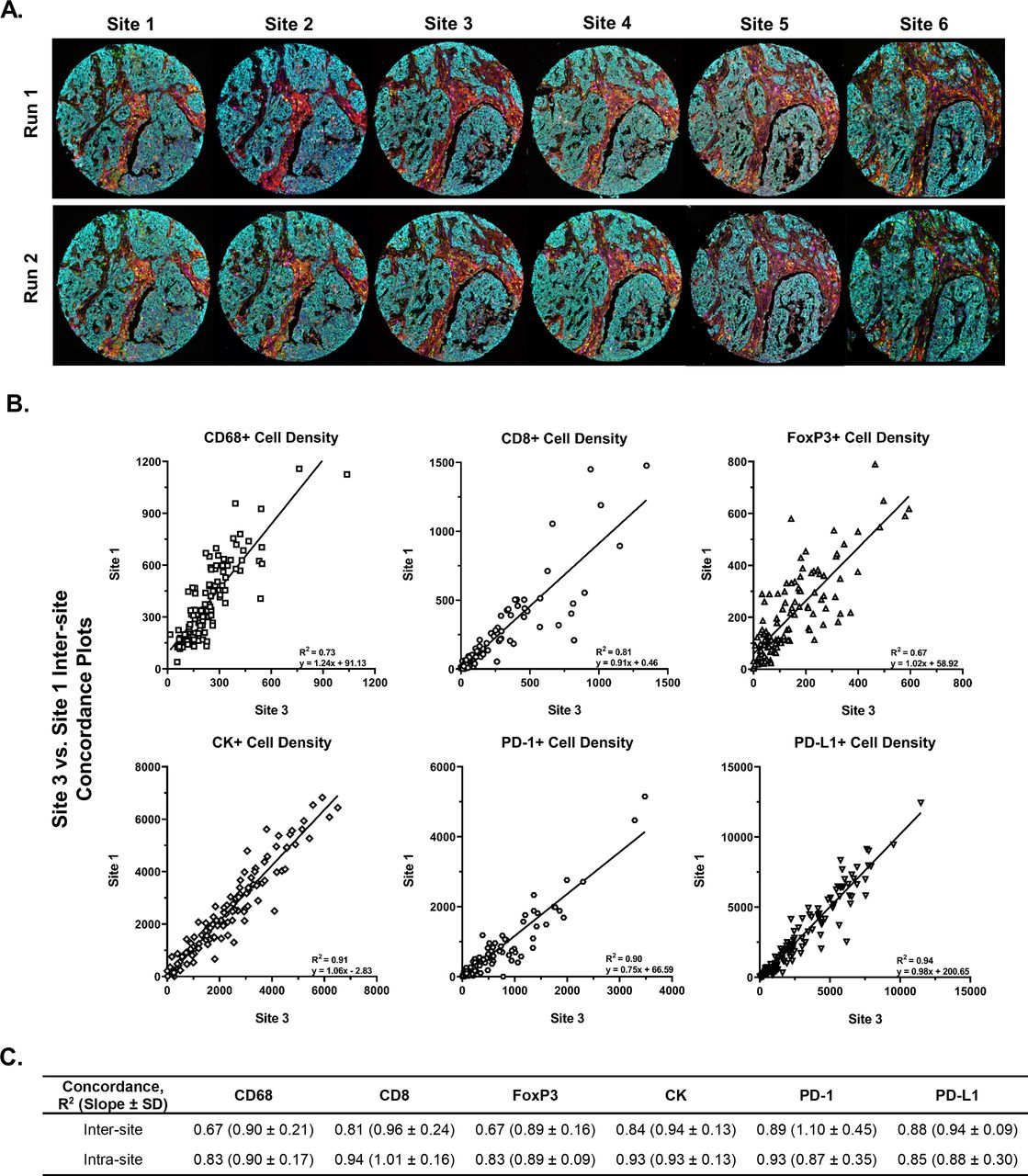
Strong intersite and intrasite concordance was observed for the cell lineages markers assessed in breast carcinoma TMA. (A) A breast carcinoma TMA was cut into 12 serial sections. Two slides were provided to each of the six sites, with one slide stained each of 2 days at each site. Images show the serial sections from a representative TMA core stained at each site over 2 days and highlight the visual consistency of automated mIF assay staining results. (B) Representative intersite cell density concordance plots for each marker, CD68, CD8, FOXP3, PD-1, PD-L1, and CK (tumor cells). The remaining intersite and intrasite comparisons are shown in online supplemental figure S2. (C) Average intersite and intrasite concordance plots densities of each cell lineage. Data shown as R2 (slope and SD of slope). The intersite and intrasite concordance results for cell lineage markers assessed in the NSCLC TMA are shown in online supplemental figure S3. P values for all concordance values are statistically significant. CK, cytokeratin; mIF, multiplex immunofluorescent ; NSCLC, non-small cell lung cancer; TMA, tissue microarray.
Intersite concordance of % PD-L1 expression by cell type and PD-1/PD-L1 proximity analysis
To demonstrate a higher level of staining reproducibility and image analysis complexity, the %PD-L1 expression by TCs and CD68+ macrophages as well as number of PD-1 cells in proximity to a PD-L1 cell were assessed. Strong concordance was observed for %PD-L1 expression by cell type, with an average fit and slope of R2=0.84 (0.91) and 0.88 (0.92) for CK+ and CD68+ in the breast TMA (figure 4A). Direct site-to-site comparison data for the breast and NSCLC TMAs are provided in online supplemental tables S2 and S3, respectively. Intersite comparison for PD-1/PD-L1 proximity using linear regression analysis showed strong fit and slope (figure 4B). The overall average intersite concordance for this analysis in the breast and lung TMAs was R2=0.82 and 0.84. Details of the R2 and slope values for each site-to-site comparison are displayed in online supplemental tables S4 and S5. Notably, the PD-1/PD-L1 proximity had stronger concordance than %PD-L1 expression at lower levels. This is of specific interest since some of the companion diagnostics used for clinical trial enrollment use a 1% cut-off for PD-L1 expression for enrollment.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Strong concordance was also achieved for %PD-L1 coexpression assessments by cell type and PD-1/PD-L1 proximity analysis. (A) Left panels: representative low and corresponding high-power photomicrographs of breast carcinoma TMA cores showing PD-L1 expression on CK+ tumor cells and CD68+ macrophages (white arrows on left and right images, respectively). Right panels: representative intersite comparison demonstrating the percent of PD-L1 displayed by CK+ and CD68+ cells. Green data points identify the two TMA cores shown in the left panels. The remaining intersite and intrasite comparisons are shown in online supplemental table S2. There was high average intersite concordance of %PD-L1 within CK+ and CD68+ cells (table shows R2 with slope and SD of slope). Similar results for intersite and intrasite concordance were observed in the NSCLC TMA and are shown in online supplemental table S3. (B) Left panel: representative image showing a TMA core with proximity map overlay, where orange dots represent PD-1+ cells, and green dots represent PD-L1+ cells. White lines display distance from all PD-L1+ cells to neighboring PD-1+ cells. Only those within 25 µm are counted (scale bar represents 200 µm). Right panel: representative intersite comparison demonstrating reproducibility of PD-1/PD-L1 proximity assessment. A high average intersite concordance for assessment of PD-1/PD-L1 proximity was observed. The individual intersite comparisons for both the breast and lung TMAs are shown in online supplemental tables S4 and S5 (table shows R2 with slope and SD of slope). P values for all concordance values are statistically significant. NSCLC, non-small cell lung cancer; TMAs, tissue microarrays.
Discussion
As immuno-oncology (IO) emerges as an effective approach to fighting cancer, quantitative immunofluorescence approaches are playing a larger role in biomarker development.17 IO brings with it the need for multivariable tests that accurately predict response and long-term benefits to patients, to help oncologists choose from the rapidly growing list of IO therapy options. Recent data suggest predictive biomarkers based on spatial arrangements of cells or coexpression patterns in FFPE tissue sections will play an important role in making IO more ‘precise’, by more accurately indicating likelihood of response to individualized treatment options.15 18 Here, we demonstrate the first steps in clinical translation of emerging multispectral imaging of multiplexed immunofluorescence (‘multispectral mIF’) technology by showing high reproducibility across six different laboratories for these key metrics.
The first step in this muli-institutional effort was the optimization of a robust, six-plex mIF assay for characterization of the PD-1/PD-L1 axis. The mIF assay described herein was performed on a Leica Bond Rx autostainer. The six-plex assay can be performed on 30 slides at a time and takes approximately 12–13 hours to perform. As such, it fits into a daily schedule that includes sample and instrument prep at the end of the day and running batches overnight, with sample imaging the subsequent day. A guiding principle behind assay optimization was that the sensitivity of the mIF panel should be quantitatively benchmarked against optimized conventional chromogenic IHC staining for each individual marker.15 16 19 20 We found that with considered selection of secondary antibodies for some of our markers, we were able to meet this standard, that is, all six stains in the mIF assay were comparable with single, chromogenic IHC stains, with the added advantage of having all the markers on a single slide.15
After this objective was achieved, we turned our focus to parameters afforded by mIF and associated slide imaging systems that are beyond the capabilities of conventional IHC approaches, including the assessment of densities of multiple markers on a single slide, determinations of spatial relationships at a single-cell level, and the quantitative evaluation of marker coexpression by individual cells. Given the growing body of evidence in this area that suggests that density and location of specific cell phenotypes within the TME,8 10 15 21 22 proximity of PD-1 to PD-L1 expression,6–8 and %PD-L1 expressed by tumor cells and/or ICs23 24 associated with response to anti-PD-1 based therapies, the expectation is that a version of the six-plex PD-1/PD-L1-axis mIF assay described herein will soon be used in collaborative oncology groups, prospective clinical trials and, ultimately, in clinical practice.
Conventional reproducibility studies focus on scoring cells as positive or negative for a given marker. Here, the reproducibility of staining intensity was assessed, which is a more rigorous metric, and one for which standard reference ranges are not currently recognized. We observed an average intersite CV for the top quartile of staining intensity of 20% compared with an average intrasite CV of 10%. We believe the relatively higher intersite variation is due to different automated stainer cleaning and maintenance protocols, the prebaking steps, and the local handling of assay reagents, for example, how accurately they are prepared or diluted; these learnings occurred after much of this reproducibility study was completed and represent a limitation of this study. Specifically, we found that baking slides at 65°C for 3 hours with a 90° rotation halfway through substantially eliminated variability between cases. Importantly, while this step was included in the TMA-based experiments, it was initiated after the intrasite and intrasite CV characterizations on tonsil tissue were performed. Notwithstanding, we believe that the data presented herein demonstrates reproducibility across sites, which will only be further improved with additional standardization of reagent, slide, and instrument handling.
Current companion and complementary PD-L1 IHC diagnostics often require pathologists to make the distinction of whether PD-L1 is expressed by a tumor cell or an IC. Pathologists have good interobserver reproducibility for the assessment of membranous %PD-L1 by tumor cells, but not for ICs, with interclass concordance metrics of >0.8 versus <0.3.25 26 This is notable, given the recent approval for the SP142 companion diagnostic assay for assessing %PD-L1 expression on ICs as a determinant for atezolizumab therapy eligibility.27–29 The mIF assay detailed herein used CD68 as marker for macrophages, that is, the majority cell type that is scored as an ‘IC’ using the PD-L1 companion diagnostic assay. Ultimately, we were able to achieve robust assessments of PD-L1 coexpression on this population. However, when first performing the intersite comparison for PD-L1 expression on the breast and NSCLC TMAs, subtle staining variability was observed across sites that affected which cells were determined to be positive or negative around the threshold. To mitigate these site-to-site differences, raw intensity values for PD-L1 expression were normalized to the three control cores in each TMA. Once these on-slide controls were used, the intersite reproducibility of %PD-L1 expression by CD68+ macrophages showed an average R2 value of 0.82, bringing it in line with %TC expression of PD-L1 by pathologists and suggesting a potential path forward for reproducible assessment of this key clinical determinant. Future studies will directly compare the predictive power of this mIF variable with pathologist visual assessments of %PD-L1 on ICs using conventional IHC.
Macrophages represent a specific image analysis challenge due to their variation in size and morphology. Here, we found that the average intersite R2 value for %PD-L1 expressed by CD68+ macrophages of 0.88 was better than the R2 value of 0.67 found when counting CD68+ cells alone. We believe this is because a % positivity calculation is a ratio (# cells positive/total # cells) rather than an absolute number (# of positive cells). As such, the value is less likely to change due to the heterogeneity of the TME between different sections and/or potential sectioning artifacts or challenges in membrane segmentation of macrophages. Along those lines, another contributing factor may be that PD-L1+ macrophages may be identified more reproducibility by the machine learning algorithm because PD-L1 expression on the membrane likely contributes to improved membrane segmentation and associated macrophage quantification. Strategies to improve membrane segmentation of macrophages that may be employed in future studies include the addition of a stain that highlights cell membranes to aid the machine learning algorithm with segmentation and/or segmenting macrophages separately from the other ICs in the TME.15
In this study, the mIF assay was performed at each of six individual locations, and the image analysis was performed at one site. The image analysis platform used in this study employs an advanced machine learning approach for segmenting and phenotyping cells. Translating mIF methods into clinical applications will most likely require creating ‘locked down’ versions of algorithms to help assure assay performance and avoid inconsistencies among laboratories. By having one site perform all the analysis with a single algorithm, we mimicked this important translational requirement. Planned future studies will address the reproducibility of the local image analysis by multiple institutions using the ‘locked-down’ algorithm that includes the aforementioned normalization to either on-slide or batch-run controls.
In summary, six laboratories collaborated to develop and optimize an automated six-plex assay focused on the PD-1/PD-L1 axis and assessed staining reproducibility. Our findings advance the current state of this assay technology by demonstrating strong intralaboratory and interlaboratory concordance for assessments of IC densities, coexpression, and proximity parameters. The approach described herein may serve as a template for assessing the analytic performance and reproducibility of emerging mIF panels for other investigative teams, with an eye toward translating such approaches into clinical trials and ultimately into the clinic.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Ethics approval
Evaluation of deidentified, archived pathology specimens described in this manuscript does not require individual patient consent. All procedures in this study were conducted in accordance with ethical principles.
Acknowledgments
The authors would like to thank the Murdock Charitable Trust, the Providence Portland Medical Foundation, the Harder Family, Robert W and Elsie Franz, Lynn and Jack Loacker, and Wes and Nancy Lematta. The authors would also like to acknowledge Justin Lucas for technical assistance during his time at BMS.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @carmenbm0404, @BernardAFox
Contributors JT, IW, ELE, DL, ERP, BAF, DLR, and CH contributed to the study concept and design, analysis and interpretation of the data, and drafting of the manuscript. JT, ELE, KR, CW, and CH developed the staining methodology. JT, ELE, KR, CW, CB-M, SMJ, JM, MJ, KR, CC, and CH were involved in data acquisition. KR performed the statistical analysis. BR and CC provided administrative and technical support. All authors read and approved the manuscript.
Funding This work was supported by National Cancer Institute 3R01CA142779-09S1A1 (JMT, DR, BF, and ERP); The MD Anderson Lung Cancer Moon Shot Program, the Cancer Prevention and Research Institute of Texas Multi-Investigator Research Award grant (RP160668), The Mark Foundation for Cancer Research (JMT), Emerson Collective (JMT); Bristol-Myers Squibb (JMT); Sidney Kimmel Cancer Center Core Grant P30 CA006973 (JMT); and The Bloomberg~Kimmel Institute for Cancer Immunotherapy.
Competing interests CB-M, SMJ, and BAF: research support from Bristol Myers Squibb II-ON program, and equipment and supply support from Akoya Biosciences; JT: research support from Bristol Myers Squibb; DLR declares that in the last 2 years, he has served as a consultant to AstraZeneca, Amgen, BMS, Cell Signaling Technology, Cepheid, Daiichi Sankyo, Danaher, GSK, Konica/Minolta, Merck, NanoString, Novartis, PAIGE.AI, PerkinElmer/Akoya Biosciences, Ultivue, and Ventana Medical Systems; BAF declares consulting for Ultivue and Neogenomics and research support from Macrogenics, Bristol Myers Squibb, Incyte, OncoSec Medical, and Merck; KR, CW, JM, CC, BR, DL, and CH: all are employees of Akoya Biosciences. No potential conflicts of interest were disclosed by the other authors.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.