Article Text

Abstract
Background Tumor metastasis is the major cause of death of colorectal cancer (CRC), and metastatic CRC remains incurable in many cases despite great advances in genetic and molecular profiling, and clinical development of numerous drugs, including immune checkpoint inhibitors. Thus, more effective treatments are urgently needed for the patients in clinical settings.
Methods We used mouse CRC metastasis models that murine Colon26 cells were subcutaneously and intravenously implanted and attempted to elucidate the tumor biological and immunological mechanisms underlying cancer metastasis. Then, we evaluated in vivo antitumor efficacy induced by agents targeting the identified molecular mechanisms using the mouse models. We validated the clinical relevancy of the findings using peripheral blood mononuclear cells obtained from stage IV metastatic CRC patients.
Results CD11b+CTLA4+ myeloid cells were systemically expanded in the metastatic settings and facilitated tumor progression and metastasis directly via generating lipid droplets in tumor cells and indirectly via inducing immune exhaustion. These events were mediated by IL1B produced via the CTLA4 signaling from the increased myeloid cells. Blocking CTLA4 and IL1B with the specific mAbs significantly suppressed tumor progression and metastasis in the mouse models resistant to anti-PD1 therapy, and the therapeutic efficacy was optimized by blocking cyclooxygenases with aspirin.
Conclusions The CD11b+CTLA4+ cells are a key driver of tumor evasion, and targeting the CTLA4-IL1B axis could be a promising strategy for treating metastatic CRC. The triple combination regimen with anti-CTLA4/IL1B mAbs and aspirin may be useful in clinical settings.
- gastrointestinal neoplasms
- immunotherapy
- myeloid-derived suppressor cells
- tumor escape
- immune evasion
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Colorectal cancer (CRC) is the third most frequently diagnosed cancer and the second most common leading cause of cancer death mostly due to metastatic relapse.1 2 Great progress in the molecular profiling of the cancer metastatic process, involving epithelial-to-mesenchymal transition (EMT) and the related cancer stem cells, has deepened understanding of the molecular landscape and has led to clinical development of many drugs targeting the signaling pathways.3 4 However, the practical improvement remains to be achieved probably due to the intratumoral biological and immunological heterogeneity produced by intrinsic and extrinsic influences in the host. Inflammation is a prominent factor to facilitate cancer metastasis and has recently re-emerged as a paramount target in cancer therapy.5 6 Proinflammatory molecules, including cyclooxygenases (COXs), prostanoids, arginase 1, and interleukin 1β (IL1B), are produced from tumor cells and myeloid cells and contribute to tumor progression and metastasis directly and indirectly via induction of immune exhaustion and dysfunction.3 7 A number of preclinical studies have shown the therapeutic efficacies of blocking these molecules using mouse tumor metastasis models,6 8 and clinical benefits of use of the small molecule inhibitors, such as aspirin9 10 and celecoxib,11 12 have been shown in patients with CRC. However, the therapeutic efficacy remains to be clinically determined in the treatment of metastatic CRC. To reinvigorate the exhausted immunity, blocking immune checkpoint pathways has been considered as a promising immunotherapy, since treatments with immune checkpoint inhibitors (ICIs), such as anti-CTLA4, anti-PD1, and anti-PDL1 mAbs, bring long-lasting durable responses in patients with metastatic cancer.13 However, the response rate remains low, and most patients eventually show acquired resistance to the treatment even if responding in the beginning.13 To predict potential responders to anti-PD1/PDL1 therapy, many biomarkers, such as PDL1 expression, mismatch repair deficiency (dMMR) and high microsatellite instability (MSI-H), have been identified using clinical samples.14 15 However, only a few patients (4%) meet the requirement according to the cumulative analysis of several phase III trials, and these are not necessarily correlated with the clinical outcomes.14 15 Combination regimens with a variety of agents, such as small molecule inhibitors and vaccines, have been now evaluated to enhance the anti-PD1/PDL1 efficacy in dozens of clinical trials.16 To date, however, many trials failed, and the evaluations are still underway. Thus, more effective treatments are urgently needed for treating CRC in the clinical settings. In this study, using the mouse CRC metastasis models, we attempted to elucidate the tumor biologic and immunologic mechanisms underlying cancer metastasis and evaluated in vivo antitumor efficacy induced by agents targeting the identified molecular mechanisms. In addition, we validated the clinical relevancy of the findings using peripheral blood mononuclear cells (PBMCs) obtained from patients with stage IV metastatic CRC.
Materials and methods
Mice
Five-week-old female BALB/c and nu/nu nude mice were purchased from Charles River Laboratories in Japan and were maintained under pathogen-free conditions until use.
Cell lines
Murine CRC Colon26 cells were purchased from Cell Resource Center for Biomedical Research at Tohoku University in Japan. Human CRC HCT116 cells were purchased from ATCC in USA. The cells were authenticated by short tandem repeat profiling and were tested for Mycoplasma negativity using a Hoechst-staining detection kit (MP Biomedicals) before use. The cells were expanded in 10% fetal bovine serum (FBS)-containing Dulbecco's Modified Eagle Medium (DMEM) (GIBCO) at 37°C and were frozen in liquid nitrogen to avoid changes occurred by a long-term culture until use. The cells were trypsinized and washed in MEMα (Wako) before use for experiments.
In vivo therapy
Mimicking patients with metastatic CRC, Colon26 cells were both subcutaneously (s.c.; 5×105 cells) and intravenously (; 2×105 cells) implanted in mice to evaluate the therapeutic efficacy on tumor growth and metastasis at the same time. Treatments were started on day 3–4 after tumor implantation (when micrometastasis was already seen in lung of the mice). Mice were orally administered with aspirin (R&D Systems) or celecoxib (Cayman Chemical) at 2 or 6 mg/kg, or PBS as a control using gavage needles daily for 5 days unless otherwise specified. As immunotherapy, mice were intraperitoneally (i.p.) injected with anti-CTLA4 mAb (Clone 9H10; BioLegend), anti-PD1 mAb (Clone 29F.1A12; BioLegend), anti-IL1B mAb (Clone B122; BioXCell), and/or mouse IgG (Clone MOPC-21; BioXCell) as a control at 5 or 10 mg/kg twice every 3–4 days. To determine the effector cells in the therapeutic mechanisms, mice were i.p. injected with anti-CD8 mAb (Clone 2.43, 200 µg; BioXCell) or anti-asialo GM1 polyclonal Ab (20 µL; BioLegend) the day before the immunotherapy. The depletion efficacy of CD3+CD8+ T cells and DX5+ NK cells (>80%) was validated by flow cytometry 1–2 days after injection. Tumor size was measured twice a week, and the volume (mm3) was calculated as follows: 0.5 × length × width.2 Two to three weeks after tumor implantation, the subcutaneous tumor, lung, and spleen were harvested from the mice for assays. Lung metastasis was assessed by counting the number of tumor nodules in lung after perfusion with water followed by fixation with Fekete’s solution. To assess systemic CTL responses in the mice, spleen cells were prestimulated with a tumor antigen gp70 peptide (1 µg/mL; MBL) for 6 days, and then the recovered CD8+ T cells (2×105 cells) were tested for cell proliferative activity in response to anti-CD3 mAb (1 µg/mL) using WST1 assay kit (Takara) and for cytotoxic activity (target=Colon26, 4 hours) using the Immunocyto Cytotoxicity Detection kit (MBL) according to the manufacturer’s instructions. Cytotoxic activity of natural killer (NK) cells was similarly assessed using Yac-1 cells as a target.
Functional analysis of CD11b+ cells
Spleen cells (SPCs) were obtained from untreated mice or the aspirin-treated mice 7 days after the last administration, and CD11b+ cells were sorted from the SPCs using a BD IMag system (BD Biosciences) with magnetic particle-conjugated antimouse CD11b mAb according to the manufacturer’s instructions (BD Biosciences). The purity (>90%) was validated by flow cytometry. The CD11b+ cells were stimulated with recombinant CD80 (10 µg/mL; R&D Systems) in the presence of anti-CTLA4 mAb or mouse IgG (1 µg/mL) for 5 days and were analyzed for intracellular IL1B expression by flow cytometry. Also, the CD11b+ cells (2×106) were cocultured with established cytotoxic T cells (CTLs) (1×106) in the presence of gp70 peptide (1 µg/mL) and/or anti-IL1B mAb, anti-CTLA4 mAb or mouse IgG as a control (1 µg/mL) for 6 days and were tested for cell proliferative activity and IFNγ productive activity using an ELISA kit (R&D Systems). In another setting, the CD11b+ cells (preinactivated with mitomycin C) were cocultured with tumor cells in the presence of anti-IL1B mAb or mouse IgG as a control for 3–5 days and were tested for cell adhesion (2 hours) using fibronectin-coated multiwell plates (Corning) and cell invasion (4 hours) using a transwell chamber with a matrigel-coated membrane (Corning) as described before.17 Lipid droplets were microscopically counted after staining with BODIPY (PMC). Tumor cells stimulated with IL1B (5 ng/mL; R&D Systems) were also used in the assay. In the in vivo setting, tumor cells (3×105) were s.c. coinjected with the CD11b+ cells (3×105) followed by intravenous injection with tumor cells (2×105) to assess the CD11b+ effect on both tumor growth and metastasis. The subcutaneous tumors were injected with anti-IL1B mAb or mouse IgG (100 µg) on day 4 after coinjection and were harvested for histological analysis on day 15. In human study, CD11b+ cells sorted from CRC patient-derived PBMCs (Proteogenex) were used in the same assays with HCT116 tumor cells. In the in vivo experiments, tumor cells (1×106) were s.c. coinjected with the CD11b+ cells (1×106) in nu/nu mice.
Flow cytometric analysis
In mouse study, cells were stained with the following antibodies after Fc blocking: anti-CD45-APC (BD Biosciences), anti-CD3e-FITC (BD), anti-CD3e-APC (BD), anti-CD3e-PerCP/Cy5.5 (BD), ant-CD8a-PE/Cy5 (BioLegend), gp70 tetramer-PE (MBL), anti-PD1-FITC (BioLegend), anti-TIGIT-PE (BioLegend), anti-CTLA4-PE (BioLegend), anti-CTLA4-PerCP/Cy5.5 (BioLegend), anti-TIM3-PE (BioLegend), anti-LAG3-FITC (Invitrogen), anti-CD11b-FITC (BioLegend), anti-Gr1-PE/Cy5 (BioLegend), anti-IL1B-PE (eBiosciences), or the appropriate isotype control. Data were acquired using the FACSCalibur cytometer (BD) and were analyzed by Cellquest software (BD). In clinical study, we used the following antibodies: anti-CD45-APC-Cy7 (BioLegend), anti-CD3-BUV496 (BD), anti-CD4-PerCP/Cy5.5 (BioLegend), anti-CD8-BUV395 (BD), anti-CD11b-BV510 (BioLegend), anti-PD1-BV605 (BioLegend), anti-CTLA4-BV785 (BD), anti-PDL1-BV785 (BD), anti-CD56-BV650 (BioLegend), anti-TIM3-APC/Cy7 (BioLegend), or the appropriate isotype control. Data were acquired using a BD LSR Fortessa X-20 cytometer (BD) and were analyzed by FlowJo software (BD). For intracellular staining, cells were treated with Cytofix/Cytoperm solution (BD) before staining. In all experiments, debris was first excluded by gating FSC/SSC, and the immunofluorescence intensity was compared with the isotype control before defining the specific molecular expression. Tumor-infiltrating immune cells were analyzed after gating CD45+ cells.
Clinical study
EDTA-added peripheral blood was collected from healthy donors (n=3) and patients with stage IV metastatic CRC (n=9; aged 57–79 years) at National Cancer Center Hospital between November 2018 and January 2020 according to the protocol approved by the Institutional Review Board of the National Cancer Center. PBMCs isolated by Ficoll (Nacalai) were analyzed by flow cytometry.
Statistical analysis
Data are presented as means±SDs unless otherwise specified. In vitro studies repeated at least three times and confirmed the reproducibility. Significant differences (p value <0.05) were statistically evaluated using GraphPad Prism V.7 software (MDF). To compare between two groups, the data were analyzed by the unpaired two-tailed Student’s t-test. To compare multiple groups, the data were analyzed by one-way analysis of variance (ANOVA), followed by the Bonferroni post hoc test for pairwise comparison of groups on the basis of the normal distributions. Non-parametric groups were analyzed by the Mann-Whitney test. In the combination therapy, significance to the single treatment was evaluated using a two-way ANOVA with Bonferroni post hoc test. Mouse survival was analyzed by Kaplan-Meier method and ranked according to the Mantel-Cox log-rank test. Correlation between two factors in clinical study was evaluated by the non-parametric Spearman’s rank test.
Results
Blocking COXs and CTLA4 synergizes in induction of antitumor immunity in mice with metastatic CRC
The representative inflammatory molecules are COXs, which produce eicosanoids such as prostaglandin E2 (PGE2) and thromboxane 2 (TXA2) from arachidonic acid, having a long history of being targeted in cancer therapy. However, the clinical efficacy remains unclear in the treatment of metastatic CRC. Also, ICIs have attracted great attention in cancer immunotherapy, while the clinical outcome is still limited to a part of the patients. Then, we first evaluated therapeutic efficacy induced by COX inhibitors and ICIs using mouse CRC metastasis models that murine CRC Colon26 cells were both subcutaneously and intravenously implanted by mimicking patients with metastatic CRC. Treatment was started on days 3–4 after tumor implantation (when micrometastasis was already observed in the lung). When we compared therapeutic efficacy between a COX1/2 inhibitor aspirin and a COX2 inhibitor celecoxib at 2 or 6 mg/kg (two cycles), potent CD8+ T cells with higher proliferative and cytotoxic activities were induced by the lower dose of aspirin, although tumor growth was significantly suppressed by both agents following increase of tumor antigen AH1-specific CD3+CD8+ T cell infiltration within the subcutaneous tumors (online supplemental figure 1). In the mice receiving the higher dose of celecoxib, spleen was extremely atrophic. Based on the results, we next compared therapeutic efficacy between the low-dose aspirin and ICIs using the same models. As well as aspirin therapy (two cycles), anti-CTLA4 therapy, but not anti-PD1 therapy, significantly suppressed tumor growth as compared with the control mice (p=0.008; figure 1A). The antitumor effect was reduced by shortening the treatment term (one cycle). However, this treatment significantly enhanced the anti-CTLA4 efficacy, particularly on mouse survival (p=0.003 vs aspirin monotherapy; figure 1B). The synergy was greatly produced by a lower dose (3 mg/kg) rather than higher dose (10 mg/kg) of anti-CTLA4 mAb (p<0.05 vs monotherapy), and potent CD8+Ki67+Gzmb+ CTLs dramatically increased within the subcutaneous tumors, although few in the monotherapy groups (figure 1C,D). These suggest that blocking COXs and CTLA4 synergizes in induction of antitumor immunity. Interestingly, CD11b+Gr1+ myeloid cells expressing CTLA4 were systemically increased in the aspirin-treated mice but were reduced by combining anti-CTLA4 therapy (figure 1E, online supplemental figure 2). This implied that the CD11b+CTLA4+ subset might be involved in the immune deterioration contributing to cancer metastasis.
Supplemental material

Blocking COXs and CTLA4 synergizes in induction of antitumor immunity in mice with metastatic colorectal cancer. BALB/c mice were both subcutaneously and intravenously implanted with murine colorectal cancer (CRC) Colon26 cells, and treatment was started on day 3 after tumor implantation. (A) Monotherapy. Mice were treated with aspirin (2 mg/kg) or PBS as a control daily on days 3–7 and days 10–14 (two cycles), or antibodies (anti-CTLA4 mAb, anti-PD1 mAb, or mouse IgG as a control at 10 mg/kg) on days 3 and 7 (n=5). (B) Combination therapy with immune checkpoint inhibitors and aspirin. Mice were treated with aspirin (2 mg/kg) daily on days 3–7 and/or antibodies (anti-CTLA4 mAb, anti-PD1 mAb, or mouse IgG at 5 mg/kg) on days 3 and 7 (n=5). (C–E) Comparison of the anti-CTLA4 dosage in combination with aspirin. Mice were treated with aspirin (2 mg/kg) daily on days 4–8 and/or anti-CTLA4 mAb (3 or 10 mg/kg) or mouse IgG (10 mg/kg) on day 4 (n=5). Tumors and spleen were harvested from the mice for assays on day 14. Tumor growth (C). T cells in the subcutaneous tumor-infiltrating cells (TILs; D). Increase of CD11b+CTLA4+ cells in spleen cells particularly of the aspirin-treated mice (immunostaining after 1-hour culture, scale=50 µm; E). *P<0.01, **p<0.05 versus control group. Graphs show means±SDs. Representative data of four independent experiments. COXs, cyclooxygenases; PBS, phosphate-buffered saline.
IL1B mediates immune exhaustion caused by the aspirin-induced CD11b+CTLA4+ cells
CTLA4 expression has been shown in human myeloid cells with immunosuppressive property.18 19 However, the characteristics of CD11b+CTLA4+ cells remain unclear in mouse settings and CRC. Then, we sorted CD11b+ cells from spleen cells (SPCs) of the aspirin-treated mice (tumor/aspirin-associated myeloid cells (TAMys)), and stimulated with a CTLA4 ligand CD80 for 5–7 days. The CTLA4+ subset was increased, but the expansion was abrogated by blocking CTLA4 with the specific mAb, suggesting requirement of the CTLA4 signaling (figure 2A). When established CTLs were cocultured with the TAMys, expressions of immune inhibitory checkpoint molecules, including PD1 and TIGIT, were markedly elevated in the CTLs (figure 2B), and the cell proliferation and IFNγ production were suppressed, suggesting induction of immune suppression and exhaustion (figure 2C). Addition of anti-CTLA4 mAb in the culture only partially rescued from the adverse events, implying other effector molecules produced from the TAMys (figure 2B,C). Inflammatory molecules derived from myeloid cells as well as tumor cells are known to damage immunity system.20 We then tested the TAMys and the cultured supernatants for various cytokines and chemokines, and finally identified IL1B that was highly expressed only in the TAMys (figure 2A). The TAMys significantly more produced active/mature IL1B in response to LPS stimulation, particularly in the presence of CD80, as compared with the control CD11b+ SPCs (online supplemental figure 3). IL1B-related IL1821 was also significantly produced from the TAMys, while the concentration was much lower than IL1B (online supplemental figure 3). NLRP3 and the complexed inflammasomes play important roles in maturation and release of IL1B.21 Indeed, IL1B production from the TAMys was significantly inhibited by addition of a potent NLRP3-specific small molecule inhibitor MCC950 in the culture (online supplemental figure 3). This suggests that NLRP3 is partly involved in the CD80-CTLA4 signaling leading to IL1B induction. Addition of anti-IL1B mAb in the culture clearly rescued from the TAMy-caused adverse events, and CTL functions were dramatically improved (figure 2B,C). These suggest that IL1B is a key effector molecule in the TAMy-induced immune suppression and exhaustion. CTL proliferation was not significantly enhanced by blocking CTLA4 despite the strong suppression of the IL1B+ subset expansion in the culture with CD80. One reason may be that it takes a longer time to recover cell cycle for cell division than protein synthesis, particularly under the inflammatory environment rich in the TAMys that potentially produce various pro-inflammatory molecules, even if each cytokine production was much less than IL1B.
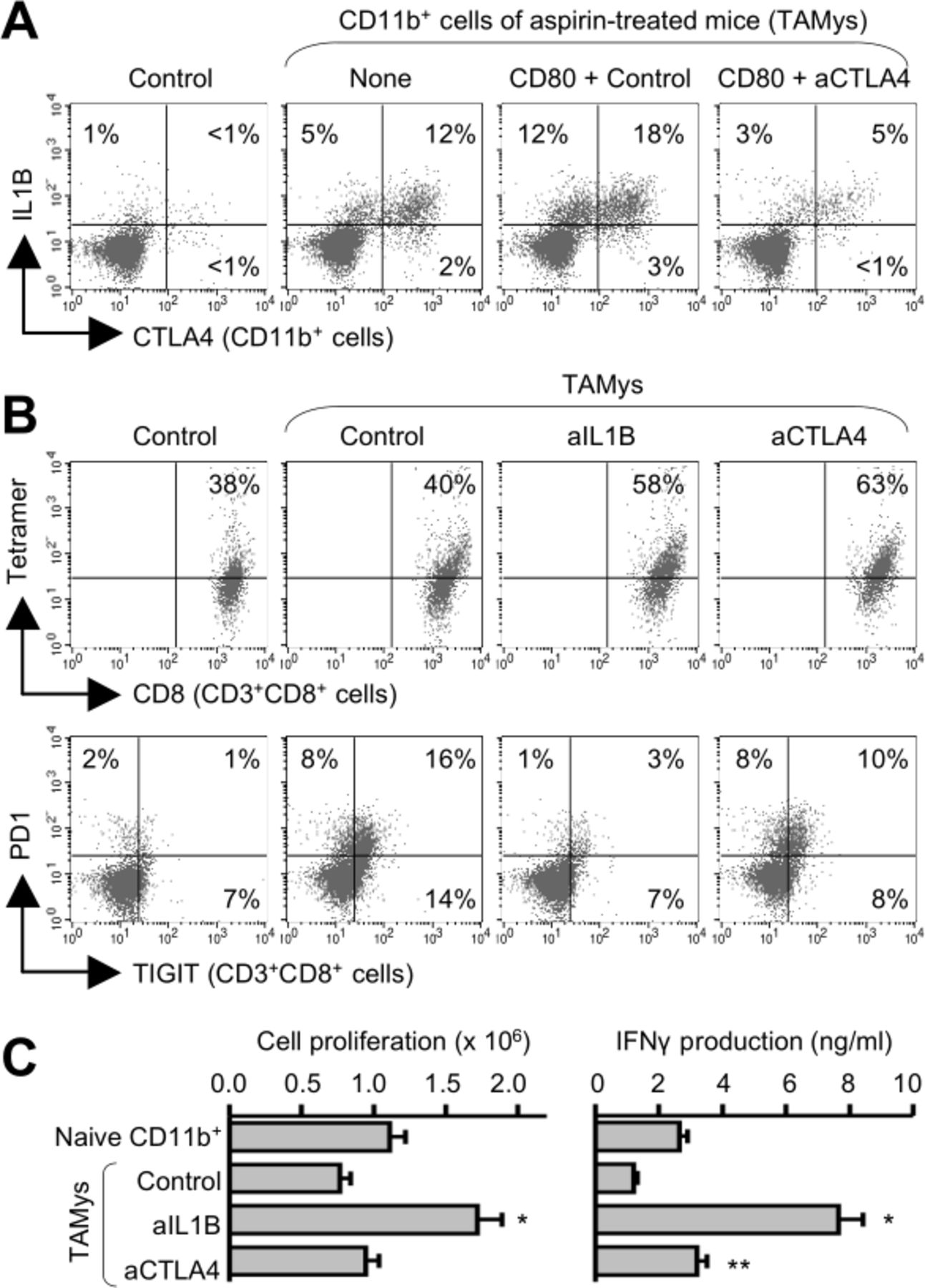
IL1B mediates immune exhaustion caused by the aspirin-induced CD11b+CTLA4+ cells. CD11b+ cells were sorted from SPCs of untreated mice (control) or aspirin-treated mice (tumor/aspirin-associated myeloid cells (TAMys)) 7 days after the last administration for assays (n=3, pooled). (A) Expansion of a CTLA4+IL1B+ subset in the TAMys via the CTLA4-CD80 axis. The CD11b+ cells were stimulated with CD80 (10 µg/mL) in the presence of anti-CTLA4 mAb or mouse IgG (1 µg/mL) for 5 days and were analyzed by flow cytometry. (B and C) Induction of potent CTLs by blocking IL1B possibly released from the TAMys. The CD11b+ cells were cocultured with established CTLs in the presence of a tumor antigen gp70 peptide and/or anti-IL1B mAb, anti-CTLA4 mAb, or mouse IgG (1 µg/mL) for 6 days. Suppression of PD1+TIGIT+ exhausted CTL induction by blocking IL1B (B). Augmentation of CTL functions in the culture with anti-IL1B mAb (n=3; means±SDs; C). *P<0.01, **p<0.05 versus control group. Representative data of three independent experiments. SPCs, spleen cells.
The TAMys facilitate tumor progression and metastasis by IL1B
We next examined how TAMys would affect tumor cells. When tumor cells were cocultured with the TAMys for 3 days, the invasive ability was significantly augmented, but the adhesive ability was significantly reduced, as compared with the control without CD11b+ cells (figure 3A). In the culture, there were many adipocyte-like cells with lipid droplets. Indeed, cells stained with a BODIPY for lipid were significantly increased in the culture with the TAMys (figure 3B). Lipid droplets are known as functional organelles responsible for aberrant lipid metabolism for survival, progression, and metastasis of cancer cells and have attracted attention as a cancer stemness.22–24 When tumor cells were coinjected with the TAMys in mice, tumor growth and metastasis were significantly facilitated following increase of lipids in the tumor tissues as compared with the control without CD11b+ cells (p=0.011; figure 3C). Anti-IL1B mAb treatment abrogated the TAMy-caused adverse events in vitro and in vivo (p<0.05; figure 3). Anti-CTLA4 treatment significantly suppressed the TAMy-caused tumor progression and metastasis (p=0.027 vs control IgG injection), and the antitumor effect was synergistically enhanced by anti-IL1B treatment (p=0.025 vs anti-CTLA4 mAb only; figure 3C). This suggests that blocking CTLA4 and IL1B restrains the TAMy activities.

The TAMys facilitate tumor progression and metastasis by IL1B. (A and B) Colon26 cells were stimulated with IL1B (5 ng/mL), or the TAMys in the presence of anti-IL1B mAb or mouse IgG (1 µg/mL) for 5 days before assays. Tumor invasion and adhesion (n=3; A). Intracellular generation of BODIPY+ lipid droplets in tumor cells (n=3; B). (C) The TAMys promote tumor progression and metastasis. Colon26 cells (3×105) were subcutaneously coinjected with CD11b+ cells (3×105) followed by intravenous injection with Colon26 cells (2×105), and 4 days later, the subcutaneous tumors were injected with anti-CTLA4 mAb and/or anti-IL1B mAb (100 µg). On day 15 after tumor implantation, tumor tissues and lungs were harvested from the mice for histological observation of tumor lipidization and metastatic tumor nodules (n=5). *P<0.01, **p<0.05 versus control group. Graphs show means±SDs. Scale=50 µm. Representative data of four independent experiments. TAMys, tumor/aspirin-associated myeloid cells.
Blocking IL1B optimizes anti-CTLA4/aspirin-induced therapeutic efficacy
We next evaluated therapeutic efficacy induced by anti-IL1B mAb in the metastasis models. Anti-IL1B therapy significantly suppressed tumor growth as compared with the control mice (p=0.004; figure 4A), and potent CTLs were systemically induced in the treated mice, while infiltration of immune cells only slightly increased in the tumors (figure 4B,C). The anti-IL1B therapy contributed to other treatments, particularly with anti-CTLA4 mAb (p<0.001 vs anti-IL1B mAb only; figure 4A). In the mice receiving anti-IL1B/CTLA4 combination therapy, antitumor effector T cells and NK cells dramatically increased in the tumor tissues (p<0.05 vs other treatments; figure 4B), and potent CTLs were systemically induced in the mice (p<0.05 vs other treatments; figure 4C). However, the therapeutic activity was lower than that induced by aspirin/anti-CTLA4 combination therapy, particularly on lung metastasis and mouse survival (p=0.023; figure 4D). The anti-IL1B/CTLA4 efficacy on tumor growth (p=0.009 vs anti-IL1B+anti-CTLA4) and mouse survival (p=0.003 vs aspirin+anti-CTLA4) was significantly enhanced by aspirin treatment (figure 4D). In the mice, infiltration of antitumor effector cells significantly increased in the subcutaneous tumors (p<0.01 vs dual combination; figure 4E), and cytotoxic activity of CTLs and NK cells was significantly augmented (p<0.05; figure 4F). Particularly, the impact of aspirin addition was much greater on NK activity rather than CTL activity, and depletion of NK cells as well as CD8+ T cells abrogated the therapeutic efficacy (p<0.001 vs no depletion; figure 4G). In the mice receiving the triple combination regimen, CD4+ cells also increased in the tumor tissues, but the percentage of Foxp3+GATA3+ regulatory T cells (Tregs) were markedly reduced in the CD4+ cell population (online supplemental figure 5). It is inferred that the CD4+ T cells indirectly contribute to the antitumor responses as helper T cells to support CTLs and NK cells. The triple combination regimen was also effective in another tumor metastasis model implanted with murine lung cancer 3LL cells (online supplemental figure 2). These results suggest that targeting the TAMys leads to successful induction of powerful antitumor immunity even under cancer metastasis.
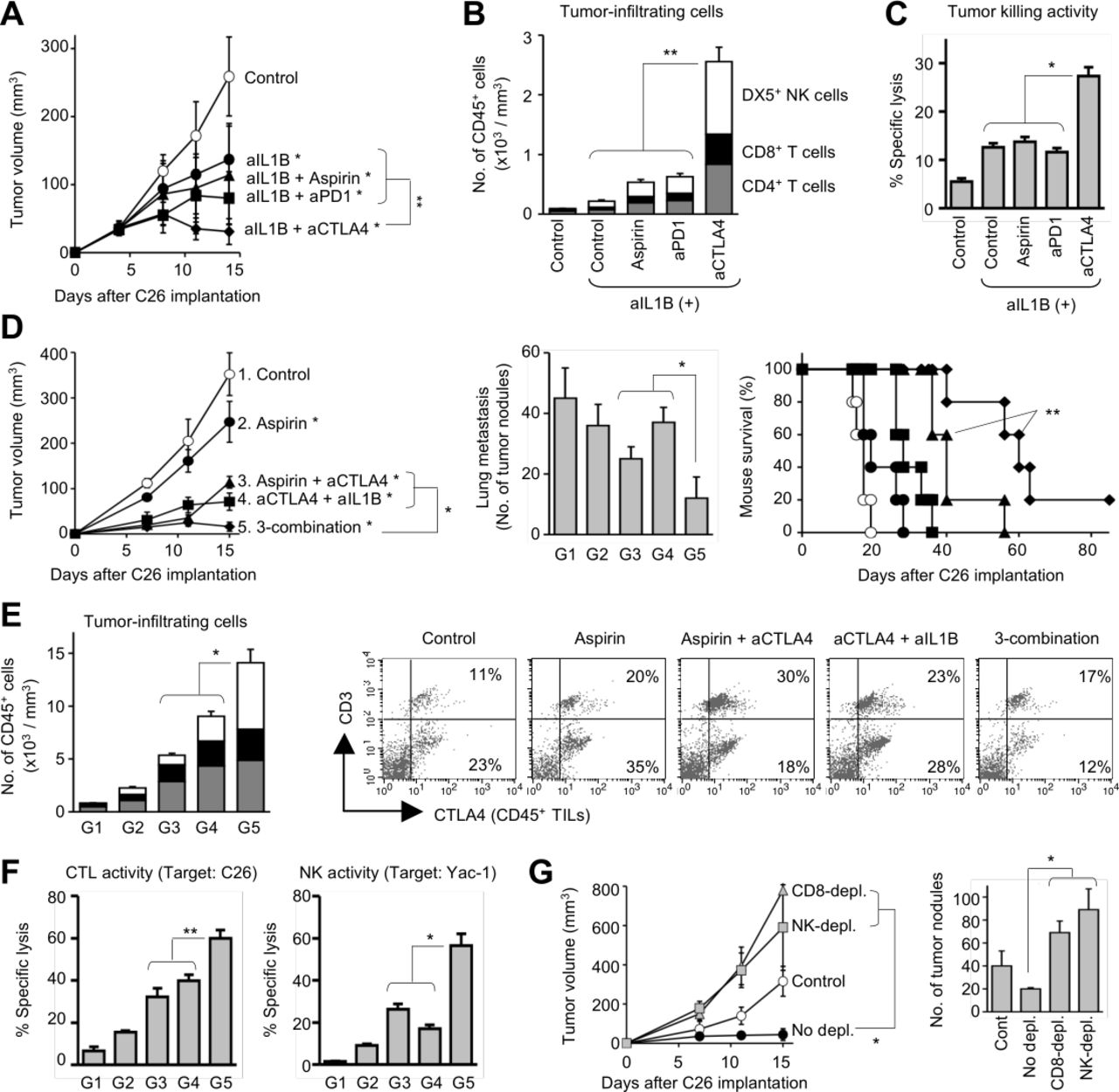
Blocking IL1B optimizes anti-CTLA4/aspirin-induced therapeutic efficacy. Mice were both subcutaneously and intravenously implanted with Colon26 cells and were treated with aspirin (2 mg/kg) daily on days 4–8 and/or antibodies (anti-CTLA4 mAb, anti-PD1 mAb, anti-IL1B, and/or mouse IgG; 5 mg/kg) on days 4 and 8. Tumors and spleen were harvested from the mice for assays on day 15. (A–C) Anti-CTLA4 therapy synergize with anti-IL1B therapy. Tumor growth (n=5; A). The number of TILs (n=5; B). Cytotoxic activity of splenic CTLs (target=Colon26, ET ratio=20:1; n=3; C). (D–G) Triple combination therapy with aspirin, anti-CTLA4 mAb, and anti-IL1B mAb. Group 1: control. Group 2: aspirin. Group 3: aspirin+anti-CTLA4 mAb. Group 4: anti-CTLA4 mAb+anti-IL1B mAb. Group 5: aspirin+anti-CTLA4 mAb+anti-IL1B mAb. Tumor growth, lung metastasis, and mouse survival (n=5; D). The number of TILs (n=5; E). Cytotoxic activity of splenic CTLs and NK cells (ET ratio=40:1; n=3; F). (G) Depletion study (n=6). Mice were intraperitoneally injected with PBS, anti-CD8 mAb, or anti-asialo GM1 ab during the triple combination therapy (days 3 and 7). *P<0.01, **p<0.05 vs control group. Graphs show means±SDs. In the stacked graphs: gray: CD3+CD4+ T cells; closed: CD3+CD8+ T cells; and open: DX5+ NK cells. Representative data of three independent experiments. ET, Effector target.
Clinical relevancy of the CD11b+CTLA4+ cells in metastatic CRC
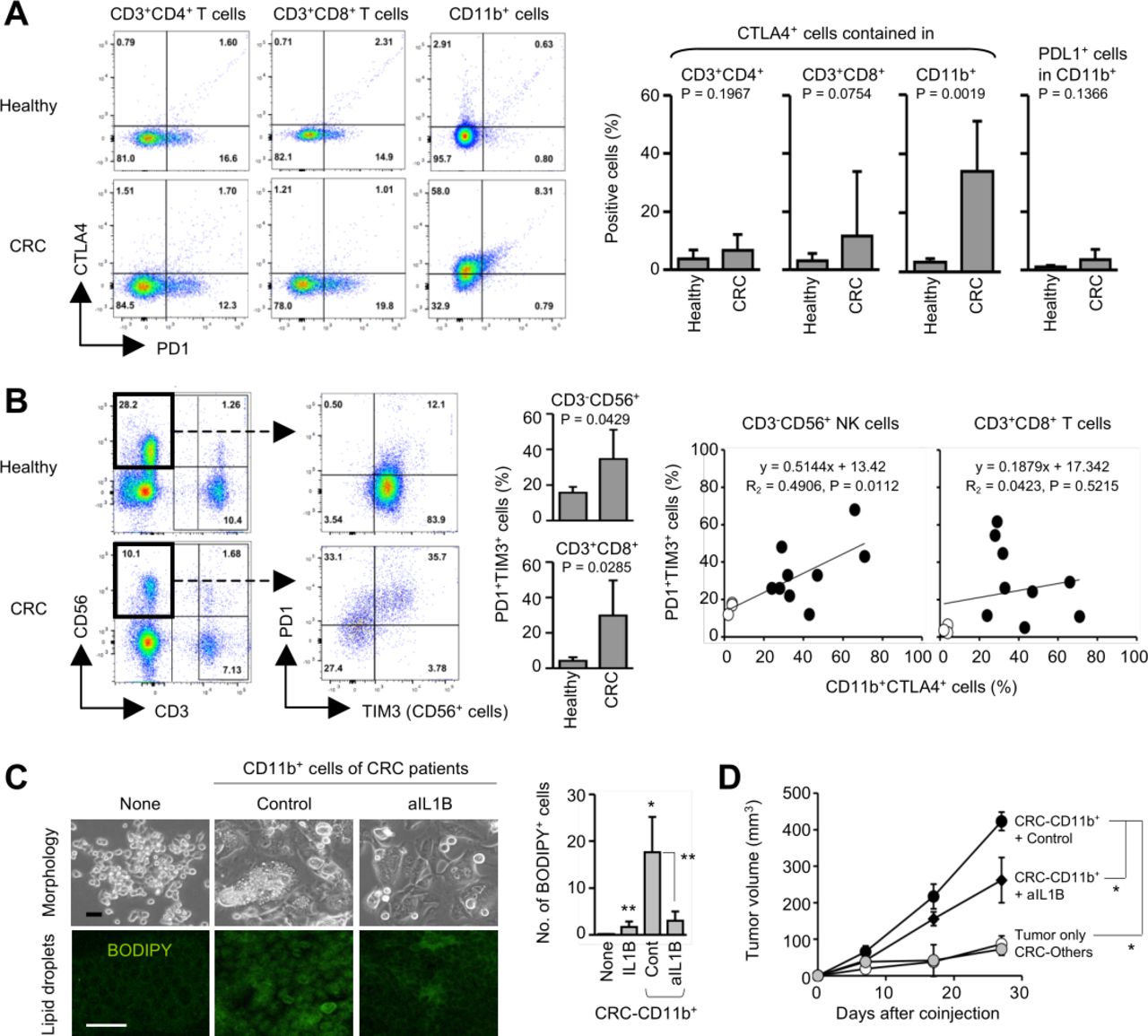
To validate the clinical relevancy of the findings, we analyzed human PBMCs obtained from patients with stage IV metastatic CRC (n=9) and healthy donors (n=3) by flow cytometry. CTLA4 was expressed in the CD11b+ myeloid cells of the patients with CRC, and the TAMy-like CD11b+CTLA4+ subset was significantly increased in the patients with CRC, but only few in healthy donors (p=0.0019; figure 5A). CTLA4 expression in CD3+CD4+ and CD3+CD8+ T cells was low or rare, and no significant differences were seen between patients with CRC and healthy donors. PDL1 is a PD1 ligand expressed in myeloid cells and tumor cells and has been targeted in cancer therapy.25 However, PDL1 was rarely expressed in the CD11b+ cells (figure 5A). These suggest that CTLA4 is a prominent marker of metastatic CRC-associated myeloid cells. In the CRC-derived PBMCs, a PD1+TIM3+ subset was significantly increased in CD56+ NK cells (p=0.0429) and CD8+ T cells (p=0.0285) as compared with those of healthy PBMCs (figure 5B). The CD11b+CTLA4+ increase was significantly correlated with the increase of the PD1+TIM3+ subset in NK cells, but not CD8+ T cells, suggesting a close relationship between them in clinical settings (p=0.0112; figure 5B). When the CRC-derived CD11b+ cells containing the CTLA4+ subset was cocultured with human CRC HCT116 cells, BODIPY+ cells with lipid droplets significantly increased (p=0.008 vs no CD11b+ cells), but addition of anti-IL1B mAb in the culture significantly suppressed the lipid generation (p=0.015; figure 5C). When HCT116 cells were coinjected with the CD11b+ cells, tumor growth was dramatically enhanced (p<0.001; figure 5D), but anti-IL1B injection significantly suppressed the tumor progression (p=0.007; figure 5D). These suggest involvement of IL1B in the mechanisms. Collectively, targeting the CTLA4-IL1B axis hopefully in combination with COXs may be a promising strategy for successfully treating metastatic CRC in clinical settings.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical relevancy of the CD11b+CTLA4+ cells in metastatic CRC. (A and B) PBMCs obtained from healthy donors (n=3) and patients with stage IV metastatic CRC (n=9) were analyzed by flow cytometry. Increase of a CTLA4+ subset in CD11b+ cells of the patients (A). Correlation between the CD11b+CTLA4+ and PD1+TIM3+ NK expansions in the patients (B). (C) Generation of lipid droplets by the CRC-derived CD11b+ cells. Human CRC HCT116 cells were cocultured with patient-derived CD11b+ cells for 5 days, and BODIPY+ lipid droplets were counted in the culture (n=3). (D) In vivo tumor progression by the patient-derived CD11b+ cells. Immunodeficient nu/nu mice were subcutaneously coinjected with HCT116 cells (1×106) and the CD11b+ cells (1×106), and the subcutaneous tumors were injected with anti-IL1B mAb or mouse IgG (100 µg) on days 4 and 11 after coinjection (n=3). *P<0.01, **p<0.05 versus control group. Graphs show means±SDs. CRC, colorectal cancer.
Discussion
Targeting inflammatory COXs has been considered as a promising strategy for treating cancer metastasis.3 7 However, at least in our metastatic CRC models, blocking COXs with aspirin was insufficient for providing significant better prognosis, although significant antitumor effects on tumor progression and metastasis were observed in the mice. We found that aspirin treatment adversely expands CD11b+CTLA4+ cells, which induce generation of lipid droplets in tumor cells and immune suppression and exhaustion in the host. IL1B produced via the CTLA4 signaling is a key effector molecule in the mechanisms. Thus, blocking CTLA4 and IL1B synergizes with aspirin treatment in successfully eliciting antitumor responses against cancer metastasis. In patients with metastatic CRC compared with healthy donors, the CD11b+CTLA4+ cells were also significantly increased in the PBMCs, and this was significantly correlated with increase of exhausted NK cells. These suggest the clinical relevancy of the findings in mouse study, and a possibility that the triple combination regimen with aspirin, anti-CTLA4 mAb and anti-IL1B mAb may be useful for treating metastatic CRC in the clinical settings.
Aspirin treatment plays an important role in empowering NK cells in the triple combination regimen. NK cells are essential for attacking disseminated tumor cells particularly in the absence of potent CTLs and MHC expression in tumor cells.26 It has been demonstrated that tumor-derived PGE2 blocks NK activation, and consequently interferes CTL activation, suggesting that NK activation is a determinant of the subsequent adaptive immunity.27 The aspirin efficacy on NK cells might be partly brought by inhibition of PGE2 produced by COX2. It should be noted that COX2 knockout upregulates COX1 expression in mice, and COX1 knockout vice versa.28 Also, COX1, but not COX2, produces TXA2 that is necessary for platelet aggregation critical for cancer metastatic process.29 These implies a risk that adverse effects may outweigh the beneficial effects in the COX2-targeted therapy potentially due to amplification of the COX1-associated events. This may account for the superiority of the aspirin efficacy to the celecoxib efficacy in our study.
Blocking CTLA4, but not PD1, greatly contributes to the aspirin therapy by suppressing reduction of the CD11b+CTLA4+ cells. CTLA4 expression that functionally interferes the CD28 signaling to activate T cells has been shown in human immunosuppressive myeloid cells, including dendritic cells (DCs)18 and mesenchymal stem cells.19 IL1B may be at least partly involved in the T cell suppression mechanisms. A study demonstrated that both aspirin and celecoxib treatments synergistically enhance anti-PD1 efficacy in several mouse tumor models.30 In the experiments, however, solid tumor models without metastasis were used, and the therapeutic efficacy was relative low despite long-term treatment (3 weeks). In clinical trials, celecoxib has been favorably used in combination with anti-PD1/PDL1 therapy, probably considering the aspirin-induced adverse effects, including gastrointestinal bleeding and adenoma.8 However, celecoxib frequently induces cardiovascular complication even if reducing the adenoma risk, and no synergized efficacy has been clinically demonstrated so far.8 Based on our results and the COX1–COX2 seesaw relationship, aspirin may be better for combining with ICIs, particularly anti-CTLA4 mAb, in the treatment of metastatic cancer. Treatment with anti-CTLA4 mAb ipilimumab, however, frequently causes high-grade autoimmunity that is largely mediated by COXs in clinical settings.31 Aspirin combination may be able to heal the anti-CTLA4-caused inflammatory responses. Interestingly, COX2 ablation in tumor cells induces de novo CTLA4 expression in many types of cancers including CRC.30 CTLA4+ tumor cells suppress maturation and functions of DCs,32 and higher CTLA4 expression in tumor tissues significantly correlates with poorer prognosis of patients with nasopharyngeal carcinoma33 and thymoma.34 Blocking COXs may be inevitably compatible with blocking CTLA4 for producing mutual benefits in treatment of metastatic CRC.
The CD11b+CTLA4+ cells induce generation of lipid droplets in tumor cells by IL1B. IL1B has been shown as a key mediator released from M2-type macrophages to induce cancer EMT.35 36 Dysregulation in lipid metabolism in cancer accelerates oncogenic signaling pathways and adversely affects the neighboring cells through the secreted components, such as lipids, cytokines, DNA, RNA, and nutrients.23 37 Intracellular lipid droplets play important roles in the aberrant lipid metabolism of cancer, although the molecular pathways of the lipid acquisition remain to be fully elucidated.38 Blockade and alternation of the lipid metabolism in cancer has attracted attention as a promising strategy for treating cancer, and a number of inhibitors targeting the molecular pathways have been clinically developed.23 37 38 Targeting IL1B may be a distinct approach to deepening the understanding of the lipid metabolism in cancer, although further study is needed to elucidate the specific molecular mechanisms.
There are many drugs targeting IL1B and the receptors, which have been clinically developed for treating inflammatory diseases such as rheumatoid arthritis and chronic obstructive pulmonary disease.39 Anti-IL1B mAb canakinumab has been now clinically evaluated in combination with immunotherapy or chemotherapy for treating various types of cancers, including lung cancer, pancreatic cancer, and renal cancer.40 Usually, anti-PD1 mAb is the first choice as an immunotherapeutic in combination with other drugs despite many failures in the clinical trials.16 However, blocking PD1 is likely helpless in the treatment of metastatic CRC, since almost no differences of PD1+ T cells in PBMCs were observed between healthy donors and patients with CRC (figure 5A), and blocking PD1 may be ineffective in suppressing tumor promotion via the TAMy-caused lipidization. Therefore, the stance should be flexibly changed on the basis of the scientific evidence, including our data. Taken together, this study may practically contribute to improvement of the clinical outcome in the treatment of metastatic CRC, although further studies are needed to elucidate the molecular mechanisms underlying the TAMy expansion and NK impairment, and the relationship with the established biomarkers, such as dMMR and MSI-H, using more clinical samples.
Data availability statement
Data are available on reasonable request.
Ethics statements
Ethics approval
Animal experiments were conducted under the approval of the Animal Care and Use Committee at the National Cancer Center Research Institute. Clinical studies were conducted under the approval of the Institutional Review Board of the National Cancer Center. Informed consent was obtained from all subjects. All activities were conducted in accordance with the ethical principles of the Declaration of Helsinki.
Acknowledgments
The authors would like to thank Takahiro Miyamoto and Hirokazu Shoji for the assistance in clinical study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors HI conducted in vitro and in vivo experiments and clinical study and wrote the manuscript. YO and MK conducted in vivo experiments. NB supervised clinical study. CK-S conceptualized, designed and directed this study, conducted experiments, analyzed data, and wrote the manuscript.
Funding This study was financially supported by the Japan Agency for Medical Research and Development AMED P-CREATE (106209 to CK-S).
Competing interests No, there are no competing interests.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.