Article Text

Abstract
Immunotherapy with gene engineered CAR and TCR transgenic T-cells is a transformative treatment in cancer medicine. There is a rich pipeline with target antigens and sophisticated technologies that will enable establishing this novel treatment not only in rare hematological malignancies, but also in common solid tumors. The T2EVOLVE consortium is a public private partnership directed at accelerating the preclinical development of and increasing access to engineered T-cell immunotherapies for cancer patients. A key ambition in T2EVOLVE is to assess the currently available preclinical models for evaluating safety and efficacy of engineered T cell therapy and developing new models and test parameters with higher predictive value for clinical safety and efficacy in order to improve and accelerate the selection of lead T-cell products for clinical translation. Here, we review existing and emerging preclinical models that permit assessing CAR and TCR signaling and antigen binding, the access and function of engineered T-cells to primary and metastatic tumor ligands, as well as the impact of endogenous factors such as the host immune system and microbiome. Collectively, this review article presents a perspective on an accelerated translational development path that is based on innovative standardized preclinical test systems for CAR and TCR transgenic T-cell products.
- Cell Engineering
- Immunotherapy, Adoptive
- Receptors, Chimeric Antigen
- Drug Evaluation, Preclinical
- T-Lymphocytes
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- Cell Engineering
- Immunotherapy, Adoptive
- Receptors, Chimeric Antigen
- Drug Evaluation, Preclinical
- T-Lymphocytes
Introduction
Adoptive T-cell therapy (ACT) represents an evolving therapeutic approach that relies on the redirection of T lymphocyte specificity toward selected tumor-associated antigens (TAAs).1 Based on early clinical observations correlating T-cell activity and infiltration with anti-tumor responses, the first ACT methods were limited to the isolation, ex vivo expansion and reinfusion of tumor-infiltrating lymphocytes, which restricted its use to a subset of patients and tumor types.2 Further development of synthetic biology and gene engineering fields prompted researchers to improve ACT products through the insertion of either chimeric antigen receptors (CARs) or transgenic T-cell receptors (TCRs), thus endowing the immune system with cytotoxic capabilities that are not naturally occurring.3 TCRs are heterodimeric glycoproteins, composed of TCR-α and β chains associated with the CD3 complex, and are able to recognize target antigens in the context of a specific peptide-major histocompatibility complex (pMHC).2 4 CARs, on the other hand, are synthetic receptors consisting of an extracellular domain containing a single-chain fragment variant (scFv), for recognition of specific tumor-antigen (without MHC-restriction), fused to an intracellular signaling region, mainly composed of the CD3z chain and costimulatory molecules such as CD28 or 4-1BB.5 Even though both approaches have shown unprecedented clinical results, several hurdles still need to be overcome to further improve long term responses and extend the success of engineered-T cells to additional hematological malignancies and hard to treat solid tumors.6–8
Broadening the clinical applicability of these treatments requires an improved understanding of the mechanisms that lead to an effective anti-tumor response. The currently available preclinical models often fail to accurately predict efficacy in the clinic due to numerous limitations, including a lack of an endogenous immune system, an inability to fully replicate the tumor microenvironment (TME) including plasticity and intratumoral heterogeneity of antigen expression, and the high costs of complicated models, among others. This lack of appropriate models, together with the difficulty in obtaining tumor biopsies from patients treated with T-cell based therapies, are limiting progress in this field. In this review, we will summarize the major achievements of engineered T-cell therapies and discuss preclinical models currently used to predict their efficacy. We will highlight the advantages and limitations of each model, with potential solutions for improvement. Whether the presented preclinical models can predict the efficacy of other ACTs, such as genetically modified gamma-delta T-cells or natural killer (NK) cells9 10 is beyond the scope of this manuscript. Preclinical models used to predict toxicity are discussed in another review as part of this same issue. Methods and tools to genetically engineer T-cells, including vectors and genome editing approaches, are beyond the scope of this review and have recently been covered elsewhere.11
CAR-T cell and TCR-T cell efficacy in clinical trials
Hematological malignancies
In the last 4 years, four autologous CD19-directed CAR-T cell products were approved in the US: Kymriah (tisagenlecleucel), Yescarta (axicabtagene ciloleucel), Tecartus (brexucabtagene autoleucel), and Breyanzi (lisocabtagene maraleucel) to treat relapsed and refractory (R/R) acute lymphoblastic leukemia (B-ALL), non-Hodgkin’s lymphoma (NHL) and Mantle Cell Lymphoma (MCL). These approvals reflect the impressive clinical results achieved by anti-CD19 CAR-T cell therapy in R/R B-cell malignancies, where complete remission (CR) rates reached 93% for B-ALL,12 13 54% for NHL14–16 and up to 67% for MCL.17 Consequently, efforts were made to extend this approach to other hematological malignancies such as multiple myeloma (MM) and acute myeloid leukemia (AML). Phase I clinical trials of anti-B-cell maturation antigen (BCMA), anti-CD123, and anti-CLL1/CD33 CAR-T cells have shown encouraging data including complete and partial responses.18 19 These efforts were recently awarded by approval of the first anti-BCMA directed CAR-T cell therapy, Abecma (idecabtagene vicleucel), for adult patients with R/R MM. Several other targets are under active clinical investigation.
Unlike CARs, TCR-T cell therapy trials are primarily targeting solid tumors, with only 16% targeting hematological malignancies.20 The most advanced TCR-T cell programs are directed against Wilms’ tumor antigen 1 (WT1) or NY-ESO-1 epitopes and have shown antileukemic activity and persistence in AML, myelodysplastic syndrome and MM, respectively.4 Autologous T-cells expressing PRAME-specific HLA-A*02:01-restricted TCR (NCT03503968) and HA-1 TCR constructs (NCT03326921) are also being tested, but no results have been reported yet.
Solid tumors
Despite the progress achieved in treating hematological malignancies, there are still no TCR-based or CAR-based T-cell products clinically approved for solid tumors. Clinical trials with TCR-engineered T-cells directed against the NY-ESO-1, MAGE, MART-1 and WT1 have been tested in a variety of cancers including melanoma, sarcoma, esophageal, and colorectal.4 Among these, NY-ESO-1-specific TCR-T cells afforded the best overall response rates in synovial sarcoma across several studies.21 Recent clinical trials testing CAR-T cells in solid tumors have most frequently focused on therapeutic products targeting MSLN, HER2, MUC-1, GD2, GPC3, EGFRvIII, and CEA in pancreatic cancer, pleural cancer, breast cancer, lung cancer, liver tumors, sarcoma, glioblastoma, and neuroblastoma.22 In most of the trials, CAR-T cell efficacy has been disappointing, although recent reports in patients with difficult to treat tumors have provided evidence for feasibility and for transient activity (including complete responses in single patients), in the absence of serious adverse events.23 24
Factors affecting clinical efficacy of engineered T-cells
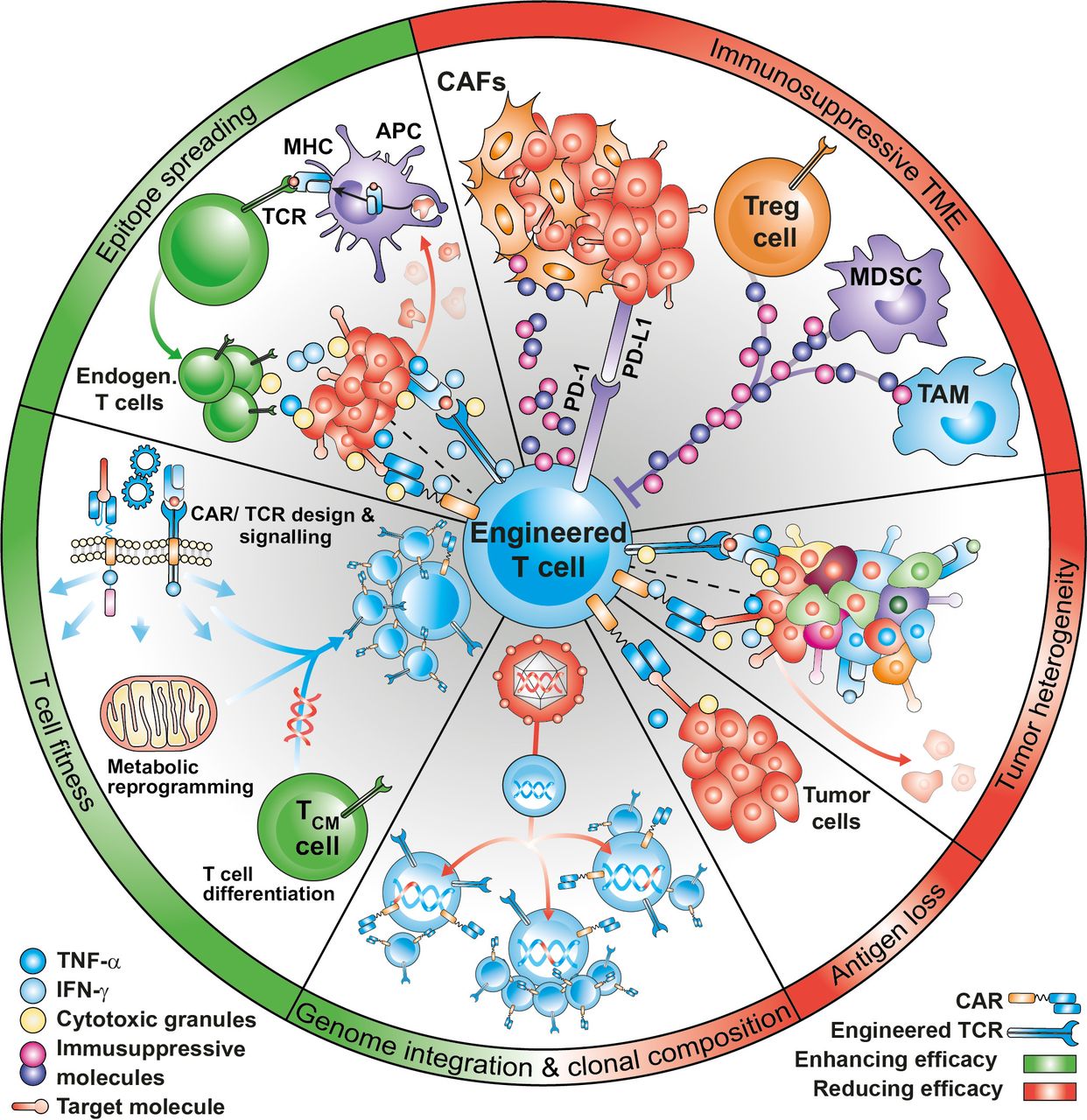
Translational clinical research has correlated the clinical response in treated patients with the expansion and persistence of infused anti-CD19-CAR-T cells.25 These two parameters have been shown to be affected by several factors, including disease histology, lymphodepleting regimens, in vivo cytokine support of the infused cells, CAR design and costimulatory domains, CAR-T cell memory phenotype and fitness, transgenic immune responses against murine origin scFv, viral integration sites in the engineered T-cells, and epitope loss or epitope masking8 (figure 1). Some of these factors were also reported in MM trials,26 whereas in AML, NHL and solid tumor studies, the lower CR rates observed could be further explained by the inability of transferred engineered T-cells to reach, infiltrate, and efficiently kill the tumor due to a highly complex and immunosuppressive TME.27 A common mechanism of resistance to CAR-T cells is the loss or downregulation of the target antigen in tumor cells. In solid tumors, heterogeneous expression of the target antigen may result in the selection of antigen low tumor variants on treatment. A possibility to overcome antigen heterogeneity would be that CAR-T cells engage the endogenous immune system to diversify the antitumor response against multiple antigens beyond the originally intended CAR or TCR target, a phenomenon known as epitope spreading. However, the inability of engineered T-cells to efficiently induce epitope spreading has emerged as a significant limitation in the context of solid malignancies.8 28
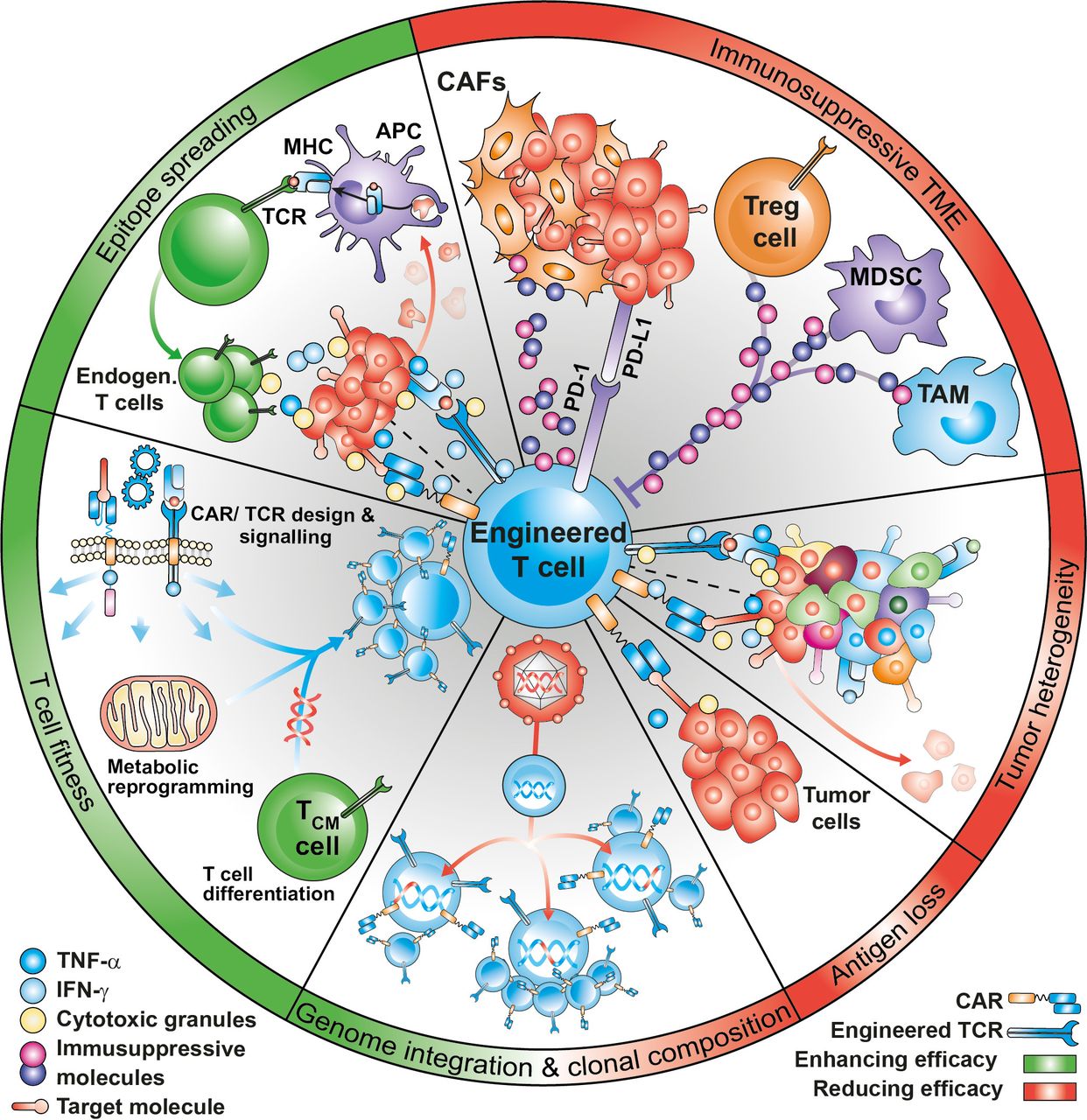
Factors affecting efficacy of engineered T cells. CAR-T or TCR-T efficacy can be influenced by several factors, that can improve (in green) or dampen (in red) clinical outcomes. The immunosuppressive tumor microenvironment and the heterogeneity and loss of antigen expression are an important causes of treatment failure. Specific baseline qualities of the infusion product, including optimal differentiation potential, metabolic profile and low expression of inhibitory molecules are key to mediate tumor control. Effective treatment with engineered T cells, especially in the context of solid tumors may require the activation of an endogenous T cell response, in a process known as epitope spreading. Finally, viral integration and clonal imbalance may have a favorable or deleterious impact on T cell efficacy. MHC, major histocompatibility complex. CAF, cancer-associated fibroblasts. APC, antigen-presenting cells. MDSC, Myeloid-derived suppressor cells. TAM, tumor-associated macrophages.
Available preclinical models and tools to assess efficacy of engineered T-cells
Currently available preclinical models have allowed for the comparison and selection of T-cell products with enhanced persistence and cytotoxic activity and were able to successfully predict the clinical efficacy of CD19-specific and BCMA-specific T-cells. However, they were unable to predict the toxicities and lack of clinical efficacy observed in many trials, especially in solid tumors. Optimization of current preclinical models to increase their predictive value is key to accelerating the development of next generation engineered T-cells. In this section, we will review the methods and tools currently used to test the efficacy of engineered T-cells, including the models able to recapitulate T-cell exhaustion, tumor heterogeneity, an immunosuppressive TME, and the impact of lymphodepletion on engineered T-cell activity (figure 2), with a focus on methods with the potential to better predict clinical outcome. Table 1 summarizes the advantages, limitations and future directions of the presented methods and tools.

{kind=link}
{kind=link}
Models and tools to assess efficacy of engineered T cells. Currently used preclinical models, including 2D cell culturing techniques and xenograft models in NSG mice, have been unable to predict the lack of responses or relapses observed in some clinical trials. Organotypic 3D models and humanized and syngeneic mouse models that better recapitulate the intra-tumor heterogeneity and the immunosuppressive TME are being developed and used in combination with novel analytical tools (at single cell level) and imaging techniques to better predict the clinical efficacy of next-generation engineered T-cell therapies. 2D, two-dimensional; MHC, major histocompatibility complex; TME, tumor microenvironment.
Methods and tools to assess efficacy of engineered T cells
CAR and TCR detection
Efficacy of engineered T-cells can be affected by the level and stability of the antigen receptor expression on the T-cell membrane. Of note, CARs are artificial by design and have as a consequence not been subjected to molecular evolution as all endogenous proteins have. As a consequence, they may not always be released to the plasma membrane but instead be retained and ultimately degraded at the level of the endoplasmic reticulum, where the biogenesis of all membrane protein takes place. Therefore, assessment of the expression of the antigen receptor in the T-cell membrane is one of the first steps in engineered-T-cell development. While the percentage and density of CAR or TCR expression can be easily assessed during T-cell manufacturing by flow cytometry, assessing the surface expression of these receptors in animal or postinfusion patient samples by flow cytometry remains challenging. In vivo tracking methods to monitor engineered T-cell trafficking, biodistribution and persistence have proven fundamental to assess the safety and efficacy of cell-based therapeutics and possibly elucidate mechanisms of therapy failure. In this regard, an effective T-cell proliferative response is the best predictor of clinical efficacy.
While flow cytometry is a sensitive detection method, and is widely used during T-cell manufacturing, the main limitations include antigen-receptor downregulation occurring after antigen encounter and, for in vivo xenograft studies, background staining due to the expression of Fc receptors in murine cells. CAR detection in animal experiments and patients could be further improved by the development of anti-idiotype antibodies, such as those used to track CD19-specific CAR-T cells derived from the FMC63 clone.29 In addition to flow cytometry, genomic and transcriptomic molecular assays have enabled the detection and quantification of engineered T-cells (qPCR and ddPCR) for longitudinal tracking due to the genomic barcode provided by vector integration in the genome and the measurement of the antigen receptor expression alone (bulk or single cell RNA sequencing). In the setting of solid tumors, CAR RNA expression has been assessed in combination with its spatial localization (RNAscope, in situ hybridization) in tumor biopsies from patients treated in clinical trials.30 However, unlike flow cytometry or microscopy, these technologies do not provide any information on the presence of the antigen receptor on the T-cell membrane, or the phenotype or function of the engineered T-cells. Recently, the use of RNAscope has been integrated with multiplex staining with antibodies (eg, MultiOmyx IF assay), providing a deeper view of proteins and genetic profiling within the TME. The simultaneous detection of CAR and T-cell differentiation and activation markers (eg, inhibitory receptors, cytokines…) in tumor tissues could provide information on CAR-T cell function in animal studies and patient biopsies. In the same line, recent advances in proteogenomics allow for deep profiling of engineered T-cells by the coupling of phenotypic features and gene expression signatures.31 The main advantages and limitations of currently used methods to detect CAR and TCR expression are summarized in table 2.
Methods and tools to assess the expression of CARs and TCRs on engineered T cells
Models and tools to assess and predict CAR- and TCR-T cell efficacy
Tonic signaling and activation-induced cell death
Genetic modification of T-cells to express CARs can result in antigen-independent constitutive signaling of the CAR, also known as tonic signaling. Different combinations of scFv, hinges, transmembrane and intracellular domains can result in different levels of tonic signaling. While low levels of tonic signaling can enhance CAR-T cell function, acute tonic signaling is associated with increased activation-induced cell death (AICD) and/or accelerated differentiation and exhaustion that results in impaired antitumor effects. Tonic signaling can be detected during primary T-cell expansion in vitro (on activation through CD3 and CD28) due to differences in CAR-T cell growth patterns and phenotypes when compared with untransduced control T-cells.32 The patterns associated with tonic signaling may differ depending on the intracellular domain included in the CAR. Incorporation of the CD28 endodomain in a CAR may result in sustained T-cell proliferation in the absence of restimulation and accelerated T-cell differentiation.33 Tonic signaling mediated by 4-1BB-costimulation is associated with an increased T-cell volume (indicative of the metabolic status) and/or increased apoptosis due to T-cell overactivation that results in impaired T cell growth.34 35 A simple method to identify tonic signaling is to plot T-cell counts and volumes during primary CAR-T cell expansion using control T-cells as a reference. Any significant variations on population doublings or T-cell volume may be indicative of tonic signaling. Other methods to detect tonic signaling include: (1) assessment of apoptosis by Annexin-V and 7-AAD staining at early time points after T-cell activation; (2) assessment of activation, differentiation and inhibitory markers by flow cytometry at the end of the primary expansion and (3) detection of CAR-embedded CD3 zeta chain phosphorylation by western blot. Acute tonic signaling during T-cell expansion is predictive of poor T-cell efficacy, and therefore should be empirically tested for each CAR construct.27 28 Mitigating tonic signaling may require testing different CAR constructs, expression cassettes and/or optimizing manufacturing conditions.29–31 An improvement in this direction would be the development or optimization of computational modeling techniques that could be used in silico to predict tonic signaling based on CAR structure of new CAR designs.
In vitro evaluation of T-cell function
To assess the antitumor activity of CAR or TCR-T cells, conventional T-cell-mediated cytotoxicity assays using co-culture of human tumor cell lines and effector T-cells are the most commonly used assays in vitro. Specific target cell lysis by engineered T-cells is generally quantified by different methods such as, the chromium (51Cr)-release assay, the luciferase-mediated bioluminescence imaging assay, the impedance-based assay, and by flow cytometry. These assays are usually combined with approaches quantifying the release of effector cytokines and/or degranulation markers as a readout of target-specific T-cell activation and function of engineered T-cells.
Specifically for TCR-T engineered T cells, functional avidity, defined by the efficient concentration of exogenous peptide eliciting half-maximum activation (EC50), has emerged as a reliable functional in vitro correlate of CD8 +T cell activity.36 EC50 is determined by measuring target cell killing or cytokine secretion in peptide titration experiments and is linked to the antigen sensitivity of T-cells. Importantly, it was shown in the context of melanoma TCR T-cell therapy that high antigen sensitivity correlated with clinical response.37 In addition, the affinity and the half-life of a TCR for its pHLA-I ligand(s) were found to be predictive of antigen sensitivity and, by extension, in vivo responses.36 Although the correlation between these molecular and biological parameters often suffer outliers, it has proven reliable enough to predict the biological behavior of most HLA-I- and HLA-II-restricted TCRs. Consequently, the preclinical in vitro efficacy assessment of natural (patient- or donor-derived) and affinity-enhanced TCR candidates largely relies on such cellular and/or biophysical evaluations.38–42
While these conventional in vitro assays are of great value to discard constructs with poor tumor reactivity, they are insufficient for predicting the functional exhaustion and AICD that T-cells encounter in vivo due to repeated stimulation. These parameters can be further investigated using a prolonged, or repetitive, ‘antigen stress assay’ in which engineered T-cells are seeded at defined effector:target ratios and repeatedly exposed to freshly seeded target cells.43 44 These long-term assays may better predict the efficacy of genetically engineered T-cells but do have some caveats as the use of two-dimensional (2D) cultures do not reproduce the architecture, phenotype, and microenvironment of solid tumors or hematological malignancies. Optimizing these in vitro evaluation assays to be used in 3D co-cultures may better predict T-cell efficacy in vivo.
Live-imaging to deepen our molecular, biophysical and cell biological understanding of CAR performance and TCR performance for engineered-T cell development
Unlike CARs, TCRs are bona fide T-cell antigen receptors conferring protection during the course of natural immune responses. Accordingly, considerable efforts have been dedicated to understanding the biological and molecular properties of TCRs expressed by protective T-cells. CARs differ considerably from TCRs in terms of structural architecture and ligand binding dynamics, but share many, membrane proximal signaling pathways. Yet, the extent to which CARs ‘walk in the footsteps’ of TCRs remains to be finetuned and optimized and will depend on parameters such as receptor-ligand size, the number of signaling chains, ligand engagement kinetics, and other variables. Of note, current CAR formats lag behind TCRs in antigen recognition performance by 3 orders of magnitude.45 Technological advances will be accelerated by quantitative and correlative assessments of structural, biophysical and cell biological parameters, as well as functional output. Fluorescent microscopy offers many opportunities for these assessments, especially when employed to monitor T-cells in action with a spatio-temporal resolution ranging (1) from several microns to less than 50 nm and (2) hours to milliseconds. Both live cell and endpoint imaging approaches have already yielded important insights into TCR-T cell-tumor and CAR-T cell-tumor cell interactions, in particular when employed in a 2D coculture setting.46–48 An extension of this approach involves the use of glass-supported lipid bilayers (SLBs), which are functionalized with recombinant TAAs, HLA/peptide, and accessory proteins. They serve as surrogate target cells as they feature experimentally defined surface densities, a prerequisite for both quantitative and mechanistic measurements of CAR-T and TCR-T recognition dynamics in real time.45 49 Since SLBs allow for live cell imaging at the single molecule or single cell level, as well as for high throughput, their potential to accelerate CAR-development is considerable but remains to be demonstrated. To realize its potential, it will require broad access in R&D and expertize in biophysics and synthetic biology.
3D models
There is growing interest in the use of 3D models for preclinical efficacy assessment of engineered-T cells. Popular examples include spheroids and organoids that better recapitulate cellular and biophysical components of in vivo tumors compared with 2D models.50
Spheroids usually describe unicellular cell-line derived floating cultures and are used for proof-of-concept studies validating CAR-T antigen-dependent cytotoxicity. Readouts typically assess overall spheroid size and viability in cocultures using fluorescence microscopy and impedance-based assays.51 52 Multicellular versions of spheroids have been developed to test immunotherapies in a more complex TME, however this has not yet been done for engineered-T cells.53 Organoids refer to complex multicellular cultures derived from tumor biopsies cultured ex vivo on 3D scaffolds. A major advantage is their recapitulation of many mutational and immune features of parental tumors, although some divergence occurs after extended culture.54 CAR-T cells have been shown to infiltrate inside patient-derived organoids, expand, degranulate and cytotoxically engage antigen-positive targets.55 56 While these readouts are important, additional readouts assessing the ability of CAR-T to remodel the TME would be useful. That includes the measurement of IFNγ-stimulated genes such as ICAM-1 and IL-12 which have been recently shown to be important for sustaining CAR T cell antitumoral activities.57 58
An alternative 3D model is the use of ‘organotypic’ models based on thick tumor slices to assess engineered T-cells performance ex vivo. A major advantage of human-derived tumor slices is that they mimic very closely the TME of in vivo tumors with the added benefit of keeping native structural integrity, which is not always the case in other 3D models. Using microscopy and specific fluorescent probes, engineered T-cells can be analyzed for distribution, migration and activation (Ca2+ responses) in real time. Immunostaining for caspase activation can also be used to measure target cell apoptosis.57
A common limitation of the above models is that they fail to mimic in vivo infiltration of engineered T-cells from blood vessels. Indeed, the coculture setups usually place engineered T-cells in direct contact with the target cells. Considering the abnormal vascularization of tumors and its effect on immune infiltration, this remains a significant gap to be addressed. Organoids can be successfully transplanted in mice and these new in vivo models could specifically address this limitation. It may also have the added benefit of more accurately predicting efficacy compared with traditional cell line tumor implants and patient-derived xenografts (PDX) due to better preservation of mutational and TME characteristics.55 59 However, this is not applicable to all 3D models and relies on the use of NSG mice that already possess many drawbacks.
Another solution involves the use of microfluidics, which aim to replicate in vivo culture dynamics in vitro, inside 3D models. A perfect example of this combination is a 3D tumor model based on the seeding of cancer cell lines on a decellularized porcine jejunum scaffold.60 Under microfluidic perfusion, these cancer cells give rise to tumors that model the architecture of in vivo tumors. Engineered T-cells can be added to the perfusion channel and show progressive invasion inside the 3D tumor model, where they can engage target cells. The application of microfluidics to other 3D models, such as organotypic tissue slices, would introduce ‘physiological infiltration’ in engineered-T cell studies. It could also have the added benefit of improving overall model viability.
A second drawback of the readouts in the aforementioned models is that the events of the engineered T-cell journey (eg, migration, activation, proliferation, degranulation, and cytotoxicity) are usually observed at specific time points. An improvement would be to observe them in real time, which has been done for CAR-T degranulation and target cell cytotoxicity in mouse models using two-photon microscopy.61
3D models offer many prospects for preclinical assessment of engineered-T cell efficacy; however, many gaps still need to be addressed to make them as close to in vivo tumors as possible.
Ex vivo omic-wide tools for assessing efficacy of engineered T-cells
Recent technological advances, such as single-cell RNA sequencing or single-cell cytokine profiling (polyfunctionality assessment), have allowed notable progress in understanding the genomic, epigenomic, and proteomic landscape of engineered T-cells in both hematological and solid tumors, providing mechanistic insights,62 63 identifying factors involved in optimal T cell function44 64–66 as well as determinants of response.25 67 68 However, most of these studies have focused on the analysis of the therapeutic product, with no analysis of post-infusion samples. Consequently, the mechanisms that lead to optimal engineered T-cell expansion and persistence after infusion are yet to be fully understood. For example, single-cell RNA sequencing of preinfusion and postinfusion specimens for a yet non-representative number of patients demonstrated dynamic changes in T cell subsets for BCMA CAR-T cells65 and CD19 –CAR-T cells66 ranging from high glycolysis levels in the infusion products, to an intermediate proliferation and cytotoxic state and to a memory-like state at later time points. Intrinsic T-cell state variability of infusion products is associated with inter-patient variability in treatment response25 67 and infusion products with enriched fractions of exhausted CD8 T-cells failed to achieve molecular responses in a study of 24 diffuse large B cell lymphoma patients.67 Emerging novel technologies, like multiomic measurements with paired readouts for gene expression, cell surface protein marker and/or TCR sequences at single-cell resolution enable deep characterization of engineered T-cell mechanisms. For solid tumors, a novel technology that utilizes barcoding of each RNA molecule according to its spatial position within the tumor tissue69 paves the way to assess engineered T-cell responses with respect to confounding factors such as tumor microheterogeneity, on-target tumor, and on-target off-tumor effects. Such advanced ex vivo analytical tools can reveal molecular mechanisms related to efficacy and treatment response, but broad applicability is limited. High costs, complex sample preparation workflows, and generation of high-dimensional data requiring specific bioinformatics expertize restricts widespread usage. Ongoing technical innovation and standardization of lab and data analysis workflows will reduce costs over time. An optimal trade-off is achieved by combining approaches, such as utilizing unbiased protocols (eg, 10X Genomics and Illumina) for discovery and targeted gene panel protocols (eg, the nCounter technology from NanoString). Overall, advanced ex-vivo analytical tools like multiomic-wide assessment of single cells are expected to provide deep insights into molecular and cellular mechanisms related to efficacy and therapy response of engineered T-cells.
Human xenograft mouse models
In order to define the therapeutic potential as well as the limitations of engineered T-cell therapies, preclinical in vivo evaluation in murine models has become the essential validation step before regulatory approval. Most in vivo preclinical studies performed to date have used human cell line-derived xenograft (CDX) models, in which tumor cell lines are engrafted in immune-deficient mice prior to infusion of TCR- or CAR-T cells. The use of tumor cell lines previously modified to express luciferase (usually firefly luciferase, FFluc) facilitates the monitoring of tumor growth by means of bioluminescence imaging.70 The most popular mouse strain for the assessment of engineered human T cell efficacy is the immunodeficient NOD/SCID/Il2rγc-/- (NSG) mouse, which is devoid of T, B, and NK cell function and has defective dendritic cells and macrophages. The preclinical efficacy of the currently approved CD19-targeting CAR-T cell therapies was demonstrated in such NSG xenograft models of leukemia and lymphoma. These models are sufficient to assess the basic anti-tumor efficacy of engineered T-cells and allow isolation of the adoptively transferred T-cells from the blood and other tissues in order to evaluate T-cell persistence, biodistribution, and phenotype. Further, employing an in vivo ‘stress test,’ by purposefully lowering the dose of engineered CAR-T-cells to levels where the therapy begins to fail, allows for the detailed comparison of CAR-T cells with different construction design.71 72
Although the NSG mouse strain is the most widely used, other mouse strains such as Scid/Beige or Rag2-/-/Il2rγc-/- can also support human tumor and T-cell xenografts. Both strains have been used for the engraftment of human hematological (AML, ALL, MM) or solid tumor (prostate and ovarian) cell lines and modeling of CAR/TCR T cell therapy.73–75 These strains, unlike NSG mice, have functional myeloid cells (dendritic cells, macrophages) and can partially mimic (restricted by species homologies) the effects mediated by the myeloid compartment of the TME during engineered-T cell therapy.76
In addition to the conventional CDX models, some CAR-T cell efficacy studies for hematological malignancies (specifically MM) have used a human bone-like xenograft model, where tumor cells are engrafted in a tissue engineered vascularized human bone matrix.77 These engineered models can be used for a variety of tumors of the hematopoietic system, as well as bone metastasizing solid tumors, and include additional levels of complexity as they allow the tumor cells to interact with the human bone microenvironment, an interaction through which stromal cell-induced resistance can be simulated. Finally, a few studies have evaluated the efficacy of autologous engineered-T cells in PDX models.78 These models better capture the natural tumor heterogeneity. However, the use of solid tumor PDX models is complicated by allogeneic and/or xenogeneic immune responses that result in high frequency of graft versus host disease (GVHD), preventing long-term follow-up of the efficacy of engineered T-cells. Moreover, the lack of human antigen expression and/or human HLA in non-tumor tissues limits the ability to test toxicity and also provides an artificial environment where the infused gene-modified human T-cells are ‘only’ able to recognize the human tumor cells. There is no tonic signaling via the endogenous TCR to promote T-cell survival, no human cytokines to support expansion and/or proliferation, and suboptimal homing of T-cells to secondary lymphoid tissues. Therefore, improvement of humanized mice as tools are necessary for better understanding of efficacy and toxicity in clinical applications.
HSCP-humanized mouse models
Humanized mouse models, in which immunocompromised mice are engrafted with human hematopoietic stem/progenitor cells (HSPCs), support the development of a multilineage human immune system. In contrast to other preclinical models, human hematopoiesis and tumor cells may coexist in these mice, allowing the study of the clinical behavior of infused engineered T-cells in a more clinically relevant milieu.79 To date, NSG mice represent a breakthrough for hematopoietic reconstitution, supporting higher engraftment rates compared with any other strain.80 To overcome the absence of human cytokine signaling, mice harboring a knock-in of human cytokines in their respective gene loci have been generated.81–83 Among these, the humanized SGM3 mouse model is generated by transplanting HSPCs into SGM3 mice, which are triple transgenic for the expression of human stem cell factor, granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-3. The improved representation of myeloid cells in these mice, besides having been instrumental in unraveling the pathophysiology of CAR-related toxicities (eg, cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome), might be crucial for establishing a more representative human TME. As a consequence, humanized SGM3 mice have successfully been employed to test the antitumor activity of CAR-T cells, both in hematological and solid tumors.84 85 Humanized SGM3 mice also proved particularly suited to generate xeno-tolerant T-cells to be employed in long-term preclinical studies without the risk of developing xenogeneic GVHD.82 84 Significant reconstitution of human dendritic cells, monocytes/macrophages and NK cells can also be achieved in humanized NSG or NOD/SCID/Jak3null (hNOJ) mice through hydrodynamic delivery of DNA vectors encoding human cytokines such as IL-15, Flt3-L, GM-CSF, M-CSF, IL-3 and IL-4.86 87 Although this model represents a good alternative to SGM3 models, cytokines are only transiently expressed (2–3 weeks) and at supraphysiological concentrations.
While the development of humanized mice represents a crucial advance, the ability to reproduce a tumor-imprinted hematological niche in these mice still requires deeper characterization. This goal can be achieved by exploiting highly informative technologies providing unbiased, genome-wide measurement of gene expression profiles of mixed cell populations. Moreover, this model still lacks the stromal component, so novel strategies to fill this gap are urgently needed. Finally, the development of endogenous responses after engineered T-cell therapy might significantly increase efficacy, but humanized mice display poor lymph node development. However, the generation of artificial lymph nodes through tissue engineering has proven effective at recreating functional immune sites once transplanted into humanized mice.88 89
Syngeneic mouse models
Human xenograft mouse models overestimate engineered T-cell therapy efficacy, fail to predict on-target off-tumor toxicities, under-represent the impact of the host immune system, and do not reflect the complexities of the TME. Syngeneic models, which involve the use of murine T-cells and murine tumor cells in mice with an intact immune system, overcome these limitations and are of particular relevance in the solid tumor setting. Syngeneic models allow for the assessment of the impact of engineered-T cells on immune cell subsets within the TME, but also on endogenous antitumor immunity. These mouse models become instrumental in evaluating strategies aimed at modulating the TME and/or at engaging the endogenous immune system to promote epitope spreading.90 Some of these strategies include armoring engineered-T cells with immune-stimulatory molecules91–95 or blocking antibodies,96 97 as well as designing T-cells that target non-tumor cell components of the TME, such as fibroblasts or tumor-associated macrophages.98 99 Syngeneic models also recapitulate the need for lymphodepletion observed in the clinical setting, which is critical for optimal antitumor efficacy in both hematological and solid tumor settings. In fact, the use of these models has allowed to demonstrate that CAR-T cells armored with proinflammatory molecules overcome the need for preconditioning,92–95 but this proof-of-concept has yet to be proven in the clinic. Syngeneic mouse models are also useful tools to evaluate synergies with other immunotherapies such as immune checkpoint inhibitors (ICIs)63 and potential on-target off-tumor toxicities that cannot be predicted by using immunocompromised xenograft mouse models. Nevertheless, antigen-binding domains specific for human targets do not often cross-react with the corresponding murine counterparts. This limitation can be partially overcome by the use of mouse-specific antigen-binding domains or transgenic mice which express human target antigens in the cell types of interest or under the regulation of the murine promoter, although differences in expression patterns of target antigens between mouse and human may exist. In addition, since the use of murinized CARs and TCRs is required, syngeneic mouse models do not allow for the testing of the same constructs that will be used in the clinic, limiting the translation of the results obtained in these models to the clinical setting.
Although the most widely used mouse strain is C57BL/6, others such as BALB/c might be used. Caution must be taken while interpreting results as significant differences have been observed between strains, especially for toxicity.100 Finally, it is important to highlight that optimized methods for the efficient generation of murine engineered-T cells are key to facilitate the use of syngeneic models, which will contribute to a better prediction of efficacy by taking into account the role of endogenous immunity.91
Transgenic mouse models to specifically assess and predict TCR T-cell efficacy
In order to better assess the efficacy and toxicity of TCR modified T-cells, transgenic mice that express human HLA class I (HLA-A2 transgenic mice) have been developed. However, differences in the mechanisms of antigen processing and presentation between mice and humans remain a limitation. While it is possible for murine cells transfected with human HLA class I to present peptides to human T-cells (or murine cells expressing a human TCR and thus HLA-restricted), due to differences in antigen processing, the cognate peptides presented in human and mice may differ. Given the fine specificity of a TCR, altered peptides may not be recognized by the human TCR or may trigger cross-reactive non-antigen specific T-cell responses. HLA-A2Kb transgenic mice that express a hybrid MHC class I molecule have also been developed. This hybrid molecule contains the human alpha1 and alpha2 domains that interact with the human HLA-A2-restricted TCR, fused to a murine alpha3 domain, which facilitates the interaction with the murine CD8 coreceptor. HLA-A2Kb mice have been used to validate the efficacy of TCR gene modified T-cells in vivo prior to first in human application, including for WT1-TCR gene modified T-cells.101 102
T-cell migration and killing dynamics in vivo
Assessing the in vivo trafficking of the infused engineered T-cells allows for further interrogation of their kinetics and anti-tumor function. In vivo detection of engineered T-cells modified to express luciferase (ex. Renilla or Gaussia luciferase) is possible by bioluminescence imaging and allows for simultaneous dual imaging of engineered-T cells and FFluc +tumor cells.103 Positron emission tomography (PET)/single photon emission CT imaging has also been used to follow in vivo CAR-T cell localization. This is possible either by directly or indirectly (immunoPET) labeling engineered T-cells with radioactive tracers,104 105 or by transducing T-cells to express traceable reporter genes.106 107
The use of intravital two-photon microscopy in mouse models permits the analysis of migration and engineered T-cell interactions at the tumor site.58 61 Combined with a genetically encoded reporter for apoptosis in tumor cells, this strategy can establish the killing dynamics of engineered T-cells in vivo.58 61 Similarly, CAR-T cells expressing various fluorescent tags for cell signaling, such as calcium signaling, have been used to characterize functional heterogeneity within CAR T-cell populations in vivo.61 This approach has also been applied to specific CAR-T cell subsets, such as CD4 +and CD8+CAR T cells, to identify the unique and shared functional properties of each population and, ideally, help guide a rationale choice for the composition of the infusion product.58 In addition, the same study revealed that the production of IFN-γ by CAR-T cells and subsequent crosstalk with the host immune system was key to sustaining the CAR-T cell killing rate over time. Strategies designed to exploit this cellular crosstalk may help increase overall antitumor activity. One caveat of these approaches is that imaging periods are limited to a few hours of continuous imaging only, although the use of imaging chambers should offer the possibility to perform longitudinal analysis.
Allogeneic CAR-T and TCR-T cells: models to evaluate host versus graft rejection in vitro
GvH and host-versus-graft (HvG) reactions are two main issues that can limit allogeneic CAR-T cell therapeutic efficacy. HLA mismatch between donor and recipient can induce alloreactive immune reactions leading to either donor cells attacking the recipient (GvH) or, conversely, patient’s immune cells rejecting the infused product (HvG), which limits CAR-T cell engraftment and long-term anti-tumor activity.108 Consequently, strategies aiming to decrease immunogenicity such as lymphodepletion or gene editing of allogeneic engineered T-cells to either resist lymphodepleting drugs, suppress MHC expression or knock down the endogenous TCR, have been developed and tested both in vitro and in in vivo animal models.109 110 To evaluate the ability of genetic modifications to prevent alloreactivity in vitro, researchers usually perform allogeneic mixed lymphocyte reactions in the presence or absence of immunosuppressive drugs such as purine nucleotide analogs and alemtuzumab (anti-CD52 monoclonal antibody). As an example, CAR T-cells from donor A, engineered to resist these drugs, are challenged by allogeneic peripheral blood mononuclear cells (PBMCs) from donor B, then proliferation and cytokine secretion of engineered T-cells are followed by flow cytometry and ELISpot, respectively.111–113 Since T-cells lacking cell-surface HLA-I are more susceptible to NK cell lysis, overexpression of the NK-cell inhibitory receptors HLA-E or HLA-G, fused to B2M via a linker, in B2M KO T-cells confers resistance to NK-cells. Functional validation of the protective effect of these additional transgenes can be obtained by coculturing engineered T-cells with allogeneic NK cells. NK cell cytotoxicity is then evaluated with an apoptosis detection kit using flow cytometry.114
In vivo, however, defining relevant models is more challenging. To assess whether modified allogeneic CAR-T cells can evade recipient cytotoxic T lymphocytes, NSG mice engrafted with donor CAR-T cells and recipient activated allogeneic PBMCs have been widely used. Persistence of donor T-cells was generally followed by phenotypic analysis of T-cells in blood and spleen using flow cytometry.
Regulatory issues
The assays described above are not only highly relevant to address basic research questions aimed at achieving a better understanding of engineered-T cell therapies, but also to fulfill regulatory requirements when applying for clinical trials or marketing authorization. Tools that determine the activities of CAR- or TCR-T cells are instrumental to set up potency assays as part of the product’s quality. Potency assays are expected to become more and more predictive for clinical efficacy over the period of product development that is usually associated with a continuously better understanding of the product’s mode of action. Apart from quality issues, non-clinical studies are an important component of the documentation required for clinical trial application. Especially for first-in-human studies, that is, in absence of any data from patients administered with a novel type of CAR- or TCR-T cell, nonclinical data are highly relevant, not only to identify potential toxicities but also to provide proof-of-concept data, which are part of pharmacodynamics and pharmacokinetic studies. Moreover, data from in vitro and in vivo models measuring anti-tumor activity are required as rationale for the starting dose in humans.115 Also, the appropriateness of potential combinations with other drugs and immunotherapies can be studied this way. To ensure the success of engineered-T cells in first-in human studies, the limitations of the models described above highlights the need for updated regulatory guidelines to help developers overcome the challenges associated with advanced cellular therapies.116 The recent update of the EMA guideline (guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells) reflects EMA’s experience with CAR-T cells, now covering details about nonclinical and clinical testing for these products. Moreover, active dialog with engineered—T-cell developers as well as a flexible regulatory approach as established by the US Food and drug administration (FDA) are attempts to fill remaining gaps.116 117 The updated EMA guideline includes an annex on special clinical consideration on the use of CAR-T cells in hematooncology. However, the combined experience gathered to date and the available guidelines remain limited, so some aspects still need to be considered on a case-by-case basis. The emergence of new generations of CAR- and TCR-T cell products, together with their extended use in solid tumors, will bring additional challenges related to safety and efficacy, which will require further guidance to ensure successful development by companies and academia.
While competent authorities are still gathering experience to draft specific guidelines for these types of products, the most important way to obtain guidance on clinical translation of a particular product is to get direct feedback from national competent authorities through formal meetings. Toward marketing authorization in the European Union (EU), scientific advice meetings are made available. More recently, Innovation Task Force meetings can be considered for novel types of development programs. During these meetings, companies can address more general, product-independent topics.
Gaps in models and tools and future directions
Most tumors are characterized by intertumor diversity and intratumor genomic heterogeneity and instability. They are a complex ecosystem that emerge and progress under selective pressure from their TME and as a response to therapy.118 Tumor-derived cell lines have long been used to study the biological processes underlying tumors, as well as to evaluate the efficacy of new anticancer immunotherapeutics. However, not all cancer subtypes are well represented by cell line models.119 Integration of different cell lines engineered to express heterogeneous levels of antigen into microphysiological ex vivo models or in NSG animals could improve studies of T cell-tumor growth dynamics and mimic tumor escape by loss of antigen expression. Further, cell line experiments inadequately simulate the spontaneous nature of oncogenesis characterized by inherit tumor heterogeneity. Immunocompetent mouse models genetically engineered to recapitulate this phenotype could be used to identify suitable targets for T cell-mediated therapy.120 Moreover, PDX models enable the investigation of the tumor closer to its patient-specific integrity.56 Their further humanization might facilitate the engraftment of immunosuppressive or stimulating cells shaping a more native environment to analyze.
Due to immune pressure by T-cell therapy, downregulation or loss of target antigens is an emerging challenge. Clinical studies have demonstrated that events such as alternative splicing or gene deletion can lead to loss of tumor antigens, subsequently causing disease relapse.121 122 Of note, cell culture assays failed to predict these scenarios emphasizing the importance of advancing in vivo systems. Encountering immune escape will require reengineering of antigen receptors towards low antigen density activity and multi-specificity, but also methods to enable fast functional evaluation of lead candidates.123 In this regard, CAR-T cell-induced epitope spreading has been studied incompletely due to lymphopenia in xenograft models which are unable to generate antitumor antibodies as found in patients.124 125 To directly test the epitope spreading, bystander hypothesis, a tumor-mixing approach using murine CAR-T cells directed against the targeted antigen-expressing murine tumor cell line engrafted to immunocompetent mice has also been developed. This model showed the limitation of CAR-T cells to efficiently kill tumors that do not express target antigen on >90% of tumor cells and allowed the evaluation of several combined approaches to eradicate tumors. Although promising, this binary model (high levels of antigen vs no expression of the antigen) need to be improved as it remains simplistic and far from the clinical reality where tumor cells are expressing a range of levels of target antigen and do not reproduce the antigen loss exerted by CAR-T cell pressure.126
Research on cancer treatment has become focused on aspects of the TME. Intratumoral hypoxia, glucose deprivation, and lactate acidifying the TME favor tolerogenic immune responses.27 In addition, stroma cells provide various growth factors and even form physical barriers resisting chemotherapy. Immunosuppressive cells contribute to a hostile environment for tumor-specific lymphocytes as well.27 While new generations of CAR-T cells were engineered to target immunosuppressive cells or withstand these rough circumstances, their efficacy was assessed only in vitro or in syngeneic mouse models.99 127 Current xenograft models fail to mimic the full complexity of the TME. Considering the multiple layers of the tumor surroundings, models need to become specialized on these distinct aspects to address them concretely, for example, by introducing immunosuppressive cells, tumor-intrinsic physical barriers or inhibitory signals.
Recently, various studies highlighted that the gut microbiota can modulate immunotherapy outcome.128 129 Analysis of human tumor entities also revealed a distinct intratumoral microbiome composition which has been correlated with response to ICIs.130 Interestingly, a preliminary study analyzed the bacterial composition in cancer patients prior to receiving CAR-T cell treatment correlating therapeutic efficacy and toxicity with abundance of certain bacterial families.131 A study on the role of microbiota-derived soluble metabolites elucidated their potential as enhancers for CAR-T cell therapy, capable of inducing metabolic and epigenetic reprogramming.132 More data regarding intratumoral colonization and the effects of microbial metabolites are a prerequisite for designing models in which therapeutic agents such as engineered-T cells encounter this additional layer of potential tumor heterogeneity. As improvement of gnotobiotic mouse models is steadily progressing to study shaping of host immunity, future approaches might consider these to assess engineered-T cell efficacy. Hence, establishment of stable and defined consortia in a humanized gnotobiotic mouse xenograft model is required to identify beneficial communities. An exemplary consortium elicited strong CD8+ T cell-mediated antitumor immunity in melanoma.133 These findings draw attention to combining engineered T-cell therapies with approaches such as fecal microbiota transfer, specific bacterial depletion, or stimulating bacterial-derived agents.
Although lymphodepletion has been clinically established to enhance successful grafting of adoptively transferred T-cells, preclinical models do not reflect the effects of the depletion regiments appropriately. In addition, mice are not always able to be treated with the same regimens as humans (eg, influenza/cy) and testing novel approaches (eg, the addition of rituximab or an anti-CD52 antibody) may not be feasible, as the mice do not express cells with the intended targets. Thus, systematic analysis of post-lymphodepletion patient samples for soluble factors or ligands might allow for the identification of biomarkers.134 This newly identified signature could be used to re-engineer existing models to recapitulate the generated milieu in patients.
Future approaches are driven by the question of whether a single or a few biomarkers are suitable to replace the conventional preclinical testing, focusing rather on precision than on broad screening strategies. An example is the T cell-tumor cell synapse. Understanding the interplay between receptor signaling, antigen-density and activating or inhibitory ligands is an essential prerequisite to both predict and determine the efficacy of the engineered cell product in a highly context-dependent manner, which further reduces time-intensive and resource-intensive animal testing.
Engineered T-cells are an emerging branch of immunotherapy for the treatment of hematological and solid tumors. The rapidly growing body of innovations in the field demands reliable, standardized and predictive pre-clinical test systems being developed and refined at the same pace to avoid roadblocks within the pipeline. Although the current landscape of models and tools is comprehensive, more interdisciplinary and specialized effort is required to overcome their limitations and consequently the bottleneck of bench-to-bedside translation, a key mission of the academic and industry partners within T2EVOLVE.
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @SoniaGuedanPhD
ML, DA, PB, CB, PB, CJB, MC, BDA, ED, DE, BG, RG, JBH, CK-M, BL, MM, JLM, EM, CQ, MR, KR, AR-G, JRR-M, ER and MT contributed equally.
Contributors SG, MC, ED, BL, JM, MH and IM conceptualised the manuscript. All authors listed wrote and edited the manuscript. All authors have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding This project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking under grant agreement No 116026. This Joint Undertaking receives support from the European Union's Horizon 2020 research and innovation program and EFPIA. We thank Chantal Kuhn for feedback on this manuscript.
Competing interests SG is an inventor on patents related to CAR-T cell therapy, filed by the University of Pennsylvania and licensed to Novartis and Tmunity, and has received commercial research funding from Novartis and Gilead. ML is an inventor on a patent application related to CAR-T cell therapy filed by Philipps-University Marburg and the University of Würzburg. DE PhD is cofunded between the academic lab led by ED as PhD supervisor and the industrial partner Invectys. EM is a Scientific Founder and holds stock options for Quell Therapeutics, consults for Orchard Therapeutics, and is Ad hoc advisor and consultant for Gilead Sciences and GSK. MT is inventor on patent applications related to CAR T cell therapy filed by Memorial Sloan Kettering Cancer Center, New York, NY; and VU Medical Center, Amsterdam, Netherlands and licensed to industry. MH is listed as an inventor on patent applications and granted patents related to CAR T cell therapy that have been filed by the Fred Hutchinson Cancer Research Center, Seattle, WA; and the University of Würzburg, Würzburg, Germany and licensed to industry. MH is a cofounder and equity holder of T-CURX, Würzburg, Germany.
Provenance and peer review Not commissioned; externally peer reviewed.