Article Text

Abstract
Background Anti-programmed cell death protein 1 (PD-1) antibodies are now routinely administered for metastatic melanoma and for increasing numbers of other cancers, but still only a fraction of patients respond. Better understanding of the modes of action and predictive biomarkers for clinical outcome is urgently required. Cancer rejection is mostly T cell-mediated. We previously showed that the presence of NY-ESO-1-reactive and/or Melan-A-reactive T cells in the blood correlated with prolonged overall survival (OS) of patients with melanoma with a heterogeneous treatment background. Here, we investigated whether such reactive T cells can also be informative for clinical outcomes in metastatic melanoma under PD-1 immune-checkpoint blockade (ICB).
Methods Peripheral blood T cell stimulation by NY-ESO-1 and Melan-A overlapping peptide libraries was assessed before and during ICB in two independent cohorts of a total of 111 patients with stage IV melanoma. In certain cases, tumor-infiltrating lymphocytes could also be assessed for such responses. These were characterized using intracellular cytokine staining for interferon gamma (IFN-γ), tumor negrosis factor (TNF) and CD107a. Digital pathology analysis was performed to quantify NY-ESO-1 and Melan-A expression by tumors. Endpoints were OS and progression-free survival (PFS).
Results The initial presence in the circulation of NY-ESO-1- or Melan-A-reactive T cells which became no longer detectable during ICB correlated with validated, prolonged PFS (HR:0.1; p>0.0001) and OS (HR:0.2; p=0.021). An evaluation of melanoma tissue from selected cases suggested a correlation between tumor-resident NY-ESO-1- and Melan-A-reactive T cells and disease control, supporting the notion of a therapy-associated sequestration of cells from the periphery to the tumor predominantly in those patients benefitting from ICB.
Conclusions Our findings suggest a PD-1 blockade-dependent infiltration of melanoma-reactive T cells from the periphery into the tumor and imply that this seminally contributes to effective treatment.
- melanoma
- lymphocytes
- tumor-infiltrating
- biomarkers
- tumor
- immunotherapy
- programmed cell death 1 receptor
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
After the unprecedented early success of immune-checkpoint blockade (ICB) in late-stage melanoma,1 2 antibody-based monotherapies, particularly those targeting the programmed cell death protein 1 (PD-1) pathway, are showing promising results even in the adjuvant setting3 and were approved for many additional cancers.4 Despite remarkable clinical benefits experienced by some patients, this is unfortunately not the case for all.5 Thus, for improved treatment allocation, biomarkers predicting clinical benefits as well as defined modes of actions of anti-PD-1 ICB are being urgently sought.6 Serum LDH so far remains the only validated biomarker in late-stage melanoma7 8 but PD-L1 expression on tumor cells, high tumor mutational burden or MHC class II expression on melanoma cells were suggested as promising candidates.9–11 Unfortunately, PD-L1 expression is an insufficient selection criterion for treatment allocation to anti-PD-1 ICB because some patients with low/negative PD-L1–expressing tumors can also benefit.12 Additional validated non-invasive biomarkers informative for disease prognosis or patient overall survival (OS) remain elusive. However, possible candidates are frequencies of myeloid-derived suppressor cells or specific T cell subsets, which are associated with clinical outcomes after ICB.13–18 Reinvigorating the exhausted circulating T cell pool through PD-1 blockade, with respective cell numbers corresponding to pretreatment tumor burden, was suggested as a potential on-treatment biomarker for therapy response.19
Although relevant T cell epitopes from somatic mutations (neoepitopes) are often unique for each patient, shared tumor-associated antigens (TAAs) such as NY-ESO-1 or Melan-A remain important target candidates. In the past, these were identified by ourselves and others as relevant targets for T cells that were associated with anti-melanoma functionality and improved clinical outcome of patients treated with multiple modalities.20–27 The aim of the present study was to investigate whether the presence of peripheral NY-ESO-1- and Melan-A-reactive T cells may be informative for clinical outcome in patients with metastatic melanoma undergoing anti-PD-1 treatment prior to the emergence of evidence of clinical benefit.
Methods
Patients
This study was conducted in two phases in two distinct cohorts. Cohort A (n=72) served as the discovery cohort, while cohort B (n=39) was used for validation. Patients with stage IV melanoma with unresectable distant metastases at the time of blood sampling were included. Venous blood samples from patients undergoing PD-1 ICB were obtained from the participating centers in Tübingen, Dresden and Lübeck before (baseline=BL) and during (follow-up=FU) therapy. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque density gradient centrifugation, cryopreserved and stored until use. Available formalin-fixed paraffin-embedded (FFPE) tumor tissue samples were selected for immunohistochemistry (IHC) analyses from patients of both cohorts that underwent surgery (median of 36 days before the start of ICB). Fresh tumor samples were collected from selected patients with metastatic melanoma from a third cohort in Tübingen. Best overall responses were assessed based on written radiological reports but did not follow RECIST criteria. All patients gave their written informed consent for biobanking and use of biomaterials as well as clinical data for scientific evaluation. The Ethics Committee of Tübingen University Hospital approved the study (792/2016BO2 and 616/2018BO2).
Detection of TAA-reactive T cells
T cells recognizing epitopes derived from NY-ESO-1 and Melan-A after in-vitro expansion were assayed. Cells were obtained either from cryopreserved PBMC aliquots or were isolated from surgically resected metastases. The latter were dissected into small pieces to induce T cell migration into the culture medium with 1000 U/mL IL-2 (Proleukin; Novartis). PBMCs and tumor cultures were stimulated with protein-spanning overlapping peptides of NY-ESO-1 and Melan-A (1 µg/mL; PepMix; JPT Peptide Technologies, Berlin). As comparison, PBMCs were stimulated with peptides of the Influenza A-derived Matrix protein 1 (Influenza). At day 4 of culture, IL-2 (40 U/mL) was added and at day 12, cells were restimulated by adding the appropriate peptides (5 µg/mL) for 12 hours in the presence of Golgi-Plug (1:1000; BD), Golgi-STOP (1:1500; BD), and CD107a-Pacific Blue antibody (H4A3; BioLegend). Next, cell surface Fc-receptors were blocked with Gamunex (GRIFOLS), and dead cells were labeled (EMA; Biotinum). Cells were stained for surface molecules using antibodies against CD3-BV510 (UCHT1; BioLegend), CD4-Alexa 647 (RPA-T4, BioLegend) and CD8-APC-H7 (SK1, BD). Afterwards, samples were treated with a fixation/permeabilization solution (BD), followed by intracellular staining using IFN-γ-PE-Cy7 (B27; BD) and TNF-Alexa 700 (Mab11; BioLegend). Samples were immediately analyzed on an LSR II cytometer (BD).
Flow cytometry data analysis
Flow cytometry data were analyzed using FlowJo (v10.4.2 and v10.7; BD). Samples were serially gated following established gating strategies (online supplemental figure 1).24 After duplicate exclusion, CD4+ and CD8+ T cells within viable CD3+ lymphocytes were selected and analyzed separately for CD107a, IFN-γ and TNF expression. Gates were set according to non-restimulated controls. The percentage of responding T cells in the restimulated versus the control sample was assessed for each of the functional markers. Antigen-reactive T cells were defined as being present when the percentage of any functional marker in either CD4+ or CD8+ T cells within the restimulated sample was ≥2 fold higher than the percentage of the corresponding population in the control sample (online supplemental figure 2). Only patients with either NY-ESO-1-, Melan-A-, or Flu-reactive T cells were included in the study.
Supplemental material
Immunohistochemistry
For IHC analyses, available BL tissue samples were cut in four-micron thick serial sections using a rotary microtome (HM355S, Thermo Scientific). Single epitope enzymatic IHC on FFPE tissue was performed on serial sections to assess Melan-A, NY-ESO-1, and CD3 expression within the tumors. Morphometric analysis was carried out on whole slide scans acquired with a Pannoramic 250 Flash III digital slide scanner (3D Histech). Quantitative morphometry was performed using the QuPath software (v0.2.3) for whole slide image analysis.28 Melan-A and NY-ESO-1 expression was assessed as the percentage of positive tumor cells within the tumor. CD3+ tumor-infiltrating lymphocytes (TILs) were grouped in TILs in the tumor cell compartment (iTILs), and TILs in the stromal compartment (sTILs). iTILs were semi-quantitatively scored as absent, non-brisk or brisk (online supplemental figure 3).
Statistics
Presence of antigen-reactive T cells was analyzed separately for NY-ESO-1, Melan-A and Influenza. Additional prognostic factors considered were age (dichotomized after the median), sex, American Joint Committee on Cancer (AJCC) staging system M category29 (M1a or M1b vs M1c), and LDH (upper (ULN) vs lower limit of normal (LLN)).
Follow-up time was defined as the period between start of anti-PD-1 treatment to last follow-up or death. Disease-specific OS probabilities were calculated, and only deaths resulting from melanoma were considered, whereas deaths resulting from other causes were censored. Progression free survival (PFS) was defined as the period from start of PD-1 ICB until the date of progression or death. Cumulative survival probabilities were estimated according to the Kaplan-Meier method and compared using log-rank testing. p<0.05 was considered statistically significant. All statistical analyses were performed using Prism (v7.0e; GraphPad Software) or SPSS (v24; IBM). For creation of graphics additionally biorender software was employed (https://biorender.com/).
Results
Patients
Peripheral blood samples from 72 patients with stage IV melanoma receiving pembrolizumab (n=35) or nivolumab plus ipilimumab (n=37) were collected at the participating clinical sites (cohort A). Samples were obtained before (BL) and a median of 42 days (interquartile-range (IQR) 42–59.9 days) after starting ICB (FU) (n=50). Median patient age was 68 years (IQR 56.8–76 years) and 61% were men. Median overall survival time (MST) was not reached. One and two-year OS was 68.3% and 54.6%, respectively. The median PFS was 5 months. Cohort B comprised PBMC samples of 39 patients with stage IV melanoma that received either pembrolizumab/nivolumab (n=11) or nivolumab plus ipilimumab (n=28). Samples were obtained at BL and at a median of 48 days (IQR 42–60.7 days) after starting ICB (FU) (n=32). Median age of patients in cohort B was 64 years (IQR 51–73 years), 66.7% were men and MST was also not reached. One and two-year OS was 87% and 60%, respectively. Median PFS was 5.5 months. Patients’ demographic data are given in table 1. Prior and subsequent treatments are summarized in online supplemental tables 1 and 2, respectively.
Determination of functional TAA-reactive T cell profiles
NY-ESO-1- and Melan-A-reactive T cell clones present in the available PBMC samples were expanded using peptide libraries of the respective proteins and their functionality was assessed by flow cytometry (figure 1A). Fifty-seven percent of all patients in cohort A possessed NY-ESO-1- and Melan-A-reactive T cells in the blood (77% in cohort B), whereas 30% possessed only NY-ESO-1-reactive T cells (18% in cohort B), but only 3% had solely Melan-A-reactive T cells (2% in cohort B). Ten percent of patients possessed neither T cell population at BL (3% in cohort B) (figure 1B). The individual dynamics of these TAA-reactive T cell populations under PD-1 ICB are depicted in figure 1C. In 15 patients of cohort A (6 of cohort B) we observed a disappearance of either NY-ESO-1- or Melan-A-reactive T cells from the blood. In contrast, the de novo appearance of TAA-reactive T cell populations (either NY-ESO-1- or Melan-A-reactive T cells or both) were found in 13 patients of cohort A (2 of cohort B) and stable populations (ie, present at baseline and follow-up) were identified in 22 patients of cohort A (24 in cohort B).

Determination of functional TAA-reactive T cell profiles in the peripheral blood of patients with stage IV melanoma under PD-1 blockade. (A) Study design and experimental workflow. (B) Distribution of NY-ESO-1-reactive and Melan-A-reactive (dark blue), NY-ESO-1-reactive (blue) or Melan-A-reactive (light blue) T cell populations or the absence (purple) of both populations before the start of PD-1 blockade in both cohorts. (C) Proportions of circulating NY-ESO-1-reactive/Melan-A-reactive T cell populations under PD-1 blockade in either cohort. Green highlight indicates patients with a loss of at least one TAA-reactive T cell population under therapy. BL, baseline; FU, follow-up; IFN-γ, interferon gamma; PD-1, programmed cell death protein 1; TAA, tumor-associated antigen.
Functional profiles of T cells responding to TAAs correlate with OS and PFS
We determined the presence of NY-ESO-1- or Melan-A-reactive T cells at BL and tested for associations with OS and PFS under PD-1 ICB in univariate analyses of cohort A (n=72). However, neither the presence of NY-ESO-1-reactive (p=0.702), nor Melan-A-reactive T cells (p=0.708) at BL was associated with prolonged OS or PFS (online supplemental table 3).
Patient and treatment characteristics
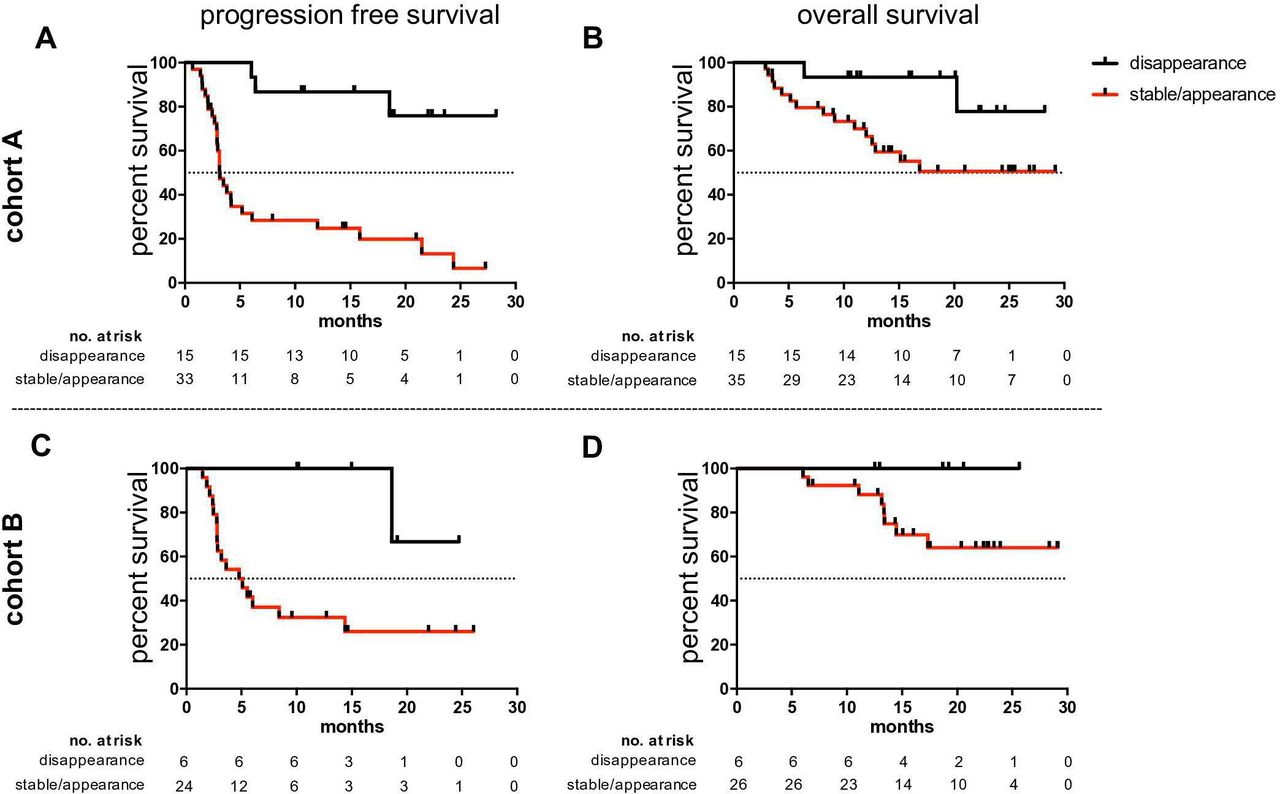
In patients with an available follow-up sample (n=50), we determined whether early alterations of the observed tumor-reactive T cell profiles under ICB (at a median of 42 days after starting therapy) were associated with OS or PFS. We found that early disappearance of TAA-reactive T cells from the circulation (whether CD4+ or CD8+ T cells, and either Melan-A- or NY-ESO-1-reactive) was significantly associated with prolonged OS (HR 0.25; 95% CI 0.09 to 0.67; p=0.045; figure 2A) and longer PFS (HR 0.13; 95% CI 0.06 to 0.27; p<0.001; figure 2B and online supplemental table 4). Median PFS was not reached for patients exhibiting a disappearance of peripheral blood TAA-reactive T cells but was only 3 months for patients in the reciprocal group. One-year OS was 93.3% for patients with disappearing TAA-reactive T cells, but only 66.5% for patients retaining or newly-acquiring such TAA-reactive T cells in the blood (two-year survival: 77.8% vs 50.6%).

Dynamics of TAA-reactive T cells under anti-PD-1 therapy correlate with clinical outcome in two independent cohorts. Disappearance of NY-ESO-1-reactive/Melan-A-reactive T cells from the circulation correlates with superior clinical outcome in two independent cohorts. Probability of overall survival (right panel) and progression-free survival (left panel) among patients with disappearing (black arm) or stable/appearing (red arm) TAA-specific T cells. Progression-free survival could not be evaluated for two patients with stable/appearing TAA-reactive T cells in cohort A and for two in cohort B. PD-1, programmed cell death protein 1; TAA, tumor-associated antigen.
The association of early disappearing NY-ESO-1-/Melan-A-reactive T cells under ICB with prolonged OS and PFS was investigated in the second, independent cohort B to validate or reject findings of cohort A. Again, we observed prolonged OS (HR 0.03; 95% CI 0.00 to 63.69; p=0.160) and PFS (HR 0.14; 95% CI 0.05 to 0.37; p=0.020) of patients with early disappearing NY-ESO-1- or Melan-A-reactive T cells under PD-1 ICB (figure 2C,D, and online supplemental table 4). Similar to cohort A, one-year OS was 100% for patients with disappearing TAA-reactive T cells, but reduced to 88.1% for patients in the reciprocal group (two-year survival: 100% vs 64.1%) (online supplemental table 4). Median PFS was also not reached for patients with the favorable phenomenon of disappearing TAA-reactive T cells while it was only 5 months for patients in the reciprocal group (online supplemental table 4). Further analysis of patients’ demographic data (table 1) revealed no significant correlations with OS or PFS (online supplemental table 5).
Combined analyses of cohort A and B also confirmed prolonged PFS (HR 0.13; 95%; CI 0.05 to 0.36; p<0.001) and OS (HR 0.21; 95% CI 0.05 to 0.91; p=0.021) in patients with early disappearances of NY-ESO-1- or Melan-A-reactive T cells (figure 3A,B, respectively). Median PFS was not reached for patients with a disappearance of TAA-reactive T cells but was only 4 months for the other patients where this was not observed. One-year OS was 95.2% for patients with disappearing TAA-reactive T cells, but only 75.9% for patients with stable or newly-appearing TAA-reactive T cells (2 year survival: 83.3% vs 56.5%) (online supplemental table 6). Similar results were obtained in a comparative analysis of the dynamics of TAA-reactive T cells in patients that received either antagonistic PD-1 antibodies alone or in combination with CTLA-4 antibodies (online supplemental figure 4).

Combined analysis of both cohorts. The disappearance of NY-ESO-1-reactive/Melan-A-reactive T cells correlates with prolonged PFS (A) and OS (B). PFS (C) and OS (D) in patients in whom both melanoma-reactive T cell populations were no longer present in the blood (black arm), or in whom just one population disappeared (orange arm), or in whom neither disappeared (red arm). OS, overall survival; PFS, progression-free survival.
A subgroup analysis stratifying the combined cohorts according to the disappearance of both, or only one of the two TAA-reactive T cell populations versus patients where this was not the case, underscored the strong correlations with clinical outcome. Thus, the subgroup of patients with a disappearance of both NY-ESO-1- and Melan-A-reactive T cells tended to have superior clinical outcomes compared with those with the disappearance of only one or neither of the relevant TAA-reactive T cell subsets (one-year-PFS: 100%, 87.5%, 27.9%; one-year survival: 100%, 93.7%, 75.9%, respectively) (figure 3C,D and online supplemental table 7).
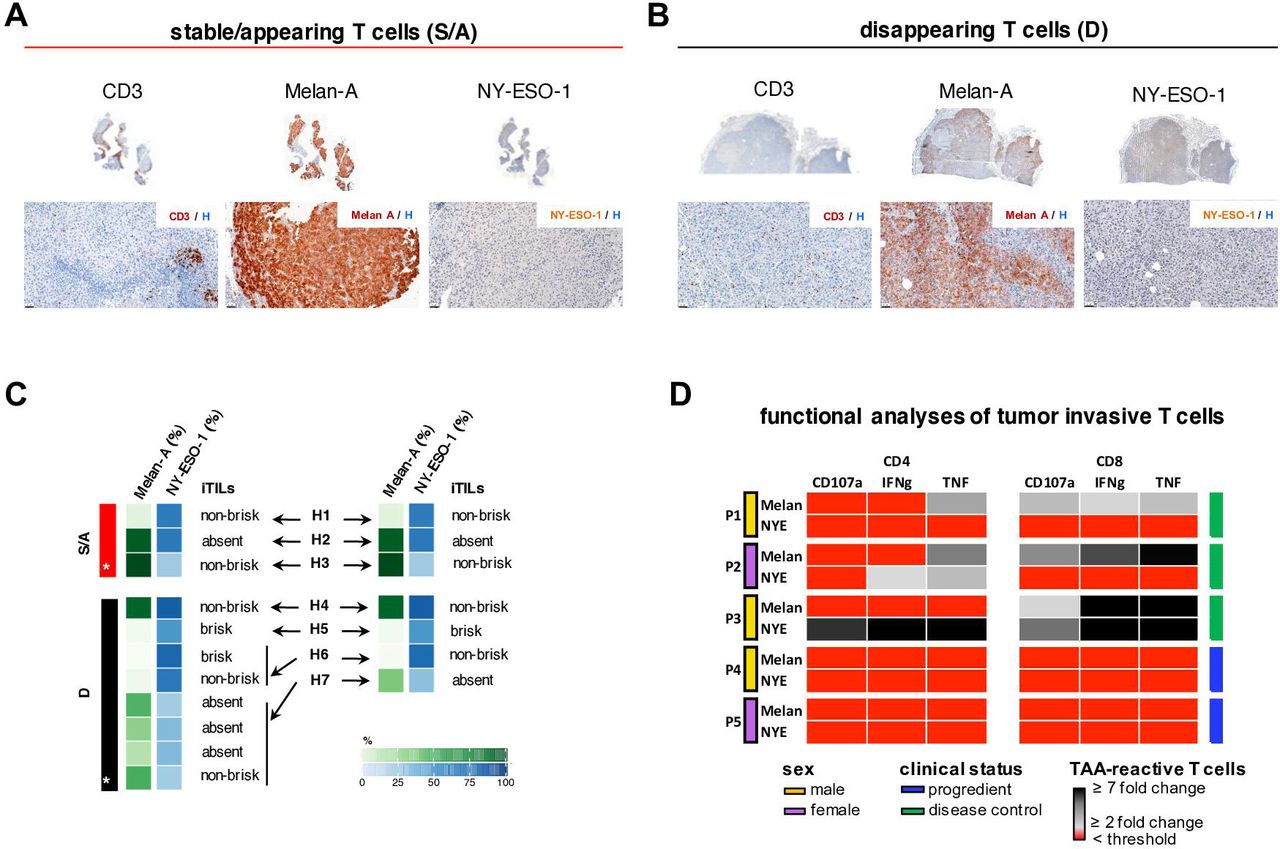
TAA expression and T cell infiltrate in metastatic tissue
T cell infiltration, as well as expression of NY-ESO-1 and Melan-A by tumor cells, was analyzed by IHC in seven patients (H1-H7) of cohort A from whom a total of 11 pre-ICB FFPE tumor tissue samples were available (patients H1-H5: 1 block per patient; patient H6: 2 tumor blocks; patient H7: 4 tumor blocks) (figure 4A–C). Overall, the proportion of NY-ESO-1-expressing tumor cells ranged between 23.3% and 83.4% (median 48%), while Melan-A-positive cells ranged between 0% and 98.8% (median 31.4%) (figure 4C). The intratumoral lymphocytic infiltrate was semi-quantitatively assessed as brisk, non-brisk, or absent on CD3-stained sections from the same tumors (figure 4C). Two of the patients from the group with stable/appearing TAA-reactive T cells in the peripheral blood under ICB showed a non-brisk tumor T cell infiltrate and in one patient we found no TILs. One patient from the group with disappearing TAA-reactive T cells showed a brisk T cell infiltrate in the tumor while two others showed a non-brisk infiltrate, and one no TILs at all. Both TAA expression and the CD3 infiltrate varied widely between the patients at BL, irrespective of the TAA-reactive T cell dynamics in the peripheral blood under ICB.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
TAA expression and infiltration of TAA-reactive T cells in metastatic tissue. Representative micrographs of cells stained for CD3, Melan-A, and NY-ESO-1 by IHC (upper row: whole tumor scans; lower row: ×200 magnification) in a patient with either stable/appearing (S/A) tumor-reactive T cells in peripheral blood (A). Representative micrographs for the same three markers (upper row: whole tumor scans; lower row: 200×magnification) in a patient with disappearing (D) tumor-reactive T cells from peripheral blood (B). Heat map depicting the percentages of Melan-A- and NY-ESO-1-positive tumor cells, as well as semiquantitative assessment of intratumoral TILs per tissue block (C, left panel) and per patient (C, right panel) in seven patients from whom histological samples were available (H1–H7) (asterisk indicates a patient for whom micrographs (A,B) are shown). Functional analyses of tumor-invasive T cell populations in five selected cases (P1–P5) (D). The presence/absence of NY-ESO-1-reactive or Melan-A-reactive CD4+ and CD8+ T cells is displayed in the heat map. Signals below the threshold are shown in red, and the fold changes above the background signal of functionally active T cells, expressing CD107a, IFN-γ or TNF, are shown in black/gray. Best overall response was assessed by imaging after surgery, usually 12 weeks later. All patients were suffering visceral metastatic disease at the time of surgery. Only in patient 5 metastases were assessed as completely resectable and surgery was performed with curative intent. Sex and disease control under ICB are depicted by the respective colors as given in the figure. S/A, stable or appearing; D, disappearing; IFN-γ, interferon gamma; iTIL, TIL in the tumor cell compartment; TAA, tumor-associated antigen.
This prompted us to further investigate TILs by addressing their specificity and functionality. The presence of NY-ESO-1- and Melan-A-reactive T cells in visceral melanoma metastases was assessed in five patients undergoing PD-1 ICB (P1-P5). The majority of tumor-infiltrating TAA-reactive T cells were CD8+, but also CD4+ T cell responses were identified. Of note, tumor infiltration by both NY-ESO-1- and Melan-A-reactive T cells was confirmed in three patients (P1-P3) showing disease control under ICB following surgery. In contrast, neither NY-ESO-1- nor Melan-A-reactive T cells could be isolated from resected tumors in two patients (P4 and P5) that suffered rapidly progressive disease in spite of ICB and surgical interventions (figure 4D). In this case series, contrary to the histological assessment, although admittedly anecdotal due to the low patient numbers, the presence of TAA-reactive T cells in these tumors did correlate with the clinical course and responses to ICB in metastatic melanoma, supporting the hypothesis that early translocation of NY-ESO-1-/Melan-A-reactive T cells from the periphery into the tumor correlated with therapy response and improved clinical outcomes under ICB.
Discussion
ICB has revolutionized medical oncology, starting with the treatment of advanced melanoma.2 However, many patients still lack treatment-related clinical responses.30 At the time of writing, there are more than 4400 clinical trials underway targeting the effects of modulating the PD-1/PD-L1 pathway,31 and much effort is being expended in investigating potential modes of action explaining clinical responses to anti-PD-1 therapy and to identify robust clinically-relevant biomarkers as correlates and predictors of response. Here we determined functional NY-ESO-1- and Melan-A-reactive T cell profiles in peripheral blood of metastatic melanoma patients receiving PD-1 ICB for non-invasive monitoring of ICB-induced effects on presumably melanoma-rejecting T cell subsets. In two independent cohorts, we observed that the early disappearance of these T cells from the peripheral blood during therapy was almost exclusively seen in patients that had a substantial benefit from PD-1 ICB. Thus, our study suggests a scenario where a superior benefit from this therapy is witnessed in a defined subset of patients in whom a therapy-induced early migration (>median of 50 days after starting therapy) of tumor-reactive T cells from the periphery into the tumor takes place.32 In this specific case, a preexisting functional NY-ESO-1-/Melan-A-reactive T cell pool in the periphery is essential (but not sufficient to predict outcome). Such an accumulation in the periphery might occur due to/as a consequence of immune cell-intrinsic and tumor microenvironment-dependent limitations33–35 and was not sufficient to discriminate patients with superior OS or PFS under ICB. These results deviate from our earlier findings in patients with heterogeneous treatment backgrounds (mostly different earlier chemotherapies), where the presence of TAA-specific T cells in the periphery did correlate with relatively prolonged OS.24 This discrepancy is likely to be caused by the impact of ICB. The MST in our previously studied cohort was 9.7 months, while MST was not reached in any of the two observed cohorts in this study, reflecting the efficacy of anti-PD-1 ICB. The presence of various other tumor-reactive T cell subsets (not monitored here), might also have been boosted by ICB and contributed to ICB-induced melanoma immunosurveillance. Several studies associate the latter with a significant increase of tumor-infiltrating T cells that is mainly observed in patients who respond to anti-PD-1 therapy.36–39 Along these lines, TILs, Melan-A and NY-ESO-1 expression patterns were determined in metastatic tissue samples taken before the start of ICB. The expression of these TAAs on tumor cells is thought to also contribute substantially to an accumulation of the respective TAA-reactive T cell subset in the peripheral blood. However, neither the presence of NY-ESO-1- and/or Melan-A-reactive T cells in peripheral blood before the start of ICB, nor their dynamics under ICB, correlated with the respective TAA expression or total T cell infiltration pattern in the available metastatic melanoma tissue samples. We assume that intra-tumor- and/or inter-metastatic heterogeneity,40 41 as well as the low number of available tissue samples, might have accounted for an incomplete picture of the tumor(s) and their microenvironment(s) determined by the histopathological assessment. We therefore studied the functional activity of NY-ESO-1- and Melan-A-reactive T cells from fresh tumor samples from patients under PD-1 ICB. Tumor infiltrating NY-ESO-1- and/or Melan-A-reactive T cells were found in all patients exhibiting disease control, while these cells were absent in those patients progressing under ICB. Thus, these results, although anecdotal due to low patient numbers, illustrate some potential advantages of functional T cell studies over purely descriptive histopathological analyses of tumor tissue slides and – even more important – already demonstrate that the monitoring of functional T cells subsets targeting certain TAAs by means of liquid biopsies allows an early identification of subgroups of patients with high chances of deriving clinical benefit from PD-1±CTLA-4 ICB.
Additionally, these data confirm the importance of NY-ESO-1 and Melan-A as target antigens of choice for immunotherapeutic approaches in melanoma. The finding that NY-ESO-1/Melan-A-reactive T cells must be detectable in the blood before therapy emphasizes the utility of these shared TAAs in future vaccination or adoptive T cell transfer trials prior to checkpoint blockade. Currently, there are several ongoing trials targeting NY-ESO-1 (NCT01967823, NCT03029273 etc.). The application of NY-ESO-1 TCR-engineered T cells has already been shown to be safe, feasible and effective in multiple myeloma patients.42 An ongoing trial administering NY-ESO-1 TCR-transduced PBMCs together with NY-ESO-1(157-165) peptide-pulsed dendritic cells and nivolumab, might help to answer this question (NCT02775292). Furthermore, immunization of advanced melanoma patients with an RNA vaccine, including NY-ESO-1, also revealed durable objective responses alone or in combination with PD-1 ICB in an exploratory analysis (NCT02410733).43
Clearly, a larger set of corresponding peripheral blood and tumor tissue samples from metastatic melanoma patients would be required to further study migration patterns of putative melanoma-rejecting T cells between the two compartments. In particular, samples of ICB responders would be most informative, but prospective study designs might be ethically problematic. Nonetheless, our model could be extended to other cancer entities and additional TAAs. The determination of such TAA-reactive peripheral T cell kinetics under therapy might help to improve ICB and increase clinical response rates by distinguishing between responders and non-responders early on during treatment.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Acknowledgments
We thank Silvia Wagner, Shannon Ottmann, Anne Mohrholz, Laura Wenke, Jenny Koop, Archana Yoganandarajah, Ulrich Schweizer, and Jürgen Winter for their support during sample collection and archiving and acknowledge the support by Open Access Publishing Fund of the University of Tübingen.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
JB and HZ contributed equally.
Correction notice Since this article was first published, Nikolaus B. Wagner has been added to the author list of this article.
Contributors JB and HZ contributed equally.
Funding This work was partially funded by Bristol-Myers Squibb (CA209-9P4), Merck Sharp & Dohme (52518), the Klaus Tschira Foundation (00.316.2017), and the Medical Faculty of the University of Tübingen (2509-0-0). Article Processing Charges were partly covered by the Open Access Publishing Fund of the University of Tübingen.
Competing interests CG reports receiving commercial research grants from Bristol-Myers Squibb, Novartis, and Roche; and is a consultant/advisory board member for Amgen, Bristol-Myers Squibb, Merck Sharp & Dohme, Novartis, and Roche. BW reports receiving commercial research grants from, is a consultant/advisory board member for, and reports receiving travel reimbursement from Bristol-Myers Squibb and Merck Sharp & Dohme. F. Meier reports receiving commercial research grants from Novartis and Roche; and has received travel support or/and speaker’s fees or/and advisor’s honoraria by Novartis, Roche, Bristol-Myers Squibb, Merck Sharp & Dohme, and Pierre Fabre. LF reported grants from the Swiss National Science Foundation, Swiss Cancer League, Hookipa Pharma, and Novartis Foundation as well as an advisory role for Novartis, Sanofi, Philogen and Bristol-Myers Squibb. PT has received speaker’s honoraria from Bristol-Myers Squibb, Novartis, Merck Sharp & Dohme, Pierre-Fabre, CureVac, Roche, Kyowa Kirin, Biofrontera, consultant’s honoraria from Bristol-Myers Squibb, Merck Serono, Novartis, Sanofi, Kirin Kyowa, and Roche and travel support from Bristol-Myers Squibb and Pierre-Fabre. SF reports receiving speaker’s fees by TAKEDA Pharmaceutical. TE has received speaker’s honoraria from Bristol-Myers Squibb, Novartis, Almiral Hermal, and Roche, consultant’s honoraria from Bristol-Myers Squibb, Merck Sharp & Dohme, Novartis, Sanofi, Pierre Fabre and Roche. TS and HN received a research grant from Novartis. GP has received speaker’s honoraria from Novartis, Roche, GlaxoSmithKline and Astellas. MWL is a co-inventor of several patents owned by Immatics Biotechnologies, and has acted as a consultant and/or advisory board member for Boehringer Ingelheim Pharma Gmbh & Co. KG. HZ is employed by CeGaT GmbH. KW-H received commercial research grants from the CatalYm GmbH and travel support from SITC (Society for Immunotherapy of Cancer). No potential conflicts of interest were disclosed by the other authors. NBW reports an advisory role for Pierre-Fabre and Sanofi, consultant's honoraria from Novartis, and has received travel support from AbbVie and Amgen outside the submitted work. KW-H is the guarantor for this manuscript.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.