Article Text

Abstract
Introduction The use of immune-checkpoint inhibitors has drastically improved the management of patients with non-small cell lung cancer (NSCLC), but innate and acquired resistances are hurdles needed to be solved. Immunomodulatory drugs that can reinvigorate the immune cytotoxic activity, in combination with antiprogrammed cell death 1 (PD-1) antibody, are a great promise to overcome resistance. We evaluated the impact of the SRC family kinases (SFKs) on NSCLC prognosis, and the immunomodulatory effect of the SFK inhibitor dasatinib, in combination with anti-PD-1, in clinically relevant mouse models of NSCLC.
Methods A cohort of patients from University Clinic of Navarra (n=116) was used to study immune infiltrates by multiplex immunofluorescence (mIF) and YES1 protein expression in tumor samples. Publicly available resources (TCGA, Km Plotter, and CIBERSORT) were used to study patient’s survival based on expression of SFKs and tumor infiltrates. Syngeneic NSCLC mouse models 393P and UNSCC680AJ were used for in vivo drug testing.
Results Among the SFK members, YES1 expression showed the highest association with poor prognosis. Patients with high YES1 tumor levels also showed high infiltration of CD4+/FOXP3+ cells (regulatory T cells (Tregs)), suggesting an immunosuppressive phenotype. After testing for YES1 expression in a panel of murine cell lines, 393P and UNSCC680AJ were selected for in vivo studies. In the 393P model, dasatinib+anti-PD-1 treatment resulted in synergistic activity, with 87% tumor regressions and development of immunological memory that impeded tumor growth when mice were rechallenged. In vivo depletion experiments further showed that CD8+ and CD4+ cells are necessary for the therapeutic effect of the combination. The antitumor activity was accompanied by a very significant decrease in the number of Tregs, which was validated by mIF in tumor sections. In the UNSCC680AJ model, the antitumor effects of dasatinib+anti-PD-1 were milder but similar to the 393P model. In in vitro assays, we demonstrated that dasatinib blocks proliferation and transforming growth factor beta-driven conversion of effector CD4+ cells into Tregs through targeting of phospholymphocyte-specific protein tyrosine kinase and downstream effectors pSTAT5 and pSMAD3.
Conclusions YES1 protein expression is associated with increased numbers of Tregs in patients with NSCLC. Dasatinib synergizes with anti-PD-1 to impair tumor growth in NSCLC experimental models. This study provides the preclinical rationale for the combined use of dasatinib and PD-1/programmed death-ligand 1 blockade to improve outcomes of patients with NSCLC.
- lung neoplasms
- tumor microenvironment
- lymphocytes
- tumor-infiltrating
- immunotherapy
- drug evaluation
- preclinical
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- lung neoplasms
- tumor microenvironment
- lymphocytes
- tumor-infiltrating
- immunotherapy
- drug evaluation
- preclinical
Introduction
The emergence of immune-checkpoint inhibitors (ICIs) for the treatment of patients with non-small cell lung cancer (NSCLC) has greatly transformed the management of this disease. The most successful strategy hitherto consists of the use of antiprogrammed cell death 1 (PD-1) antibody to enhance T-cell activation and reduce tumor immunosuppression.1 Pembrolizumab, nivolumab and atezolizumab are Food and Drug Administration (FDA)-approved antibodies to block the PD-1/programmed death-ligand 1 (PD-L1) axis in patients with advanced NSCLC as first-line or second-line therapy.1 2 However, in spite of the striking long-lasting clinical responses previously unseen with other strategies, current immunotherapy only benefits 30%–40% of the patients when applied in monotherapy, and many of the responders will develop acquired resistance over the course of the treatment.3 4 In addition, there is no reliable biomarker to predict response, with PD-L1 expression and tumor mutational burden as possible (but not completely accurate) indicators of efficacy.5 The mechanisms of resistance are not fully elucidated, but the presence of an immune desert tumor microenvironment (TME), with low infiltration of effector cytotoxic CD8+ cells, and high infiltration of regulatory T cells (Tregs) or myeloid-derived suppressor cells are cornerstones of this process.6
Because of these hurdles, novel strategies are actively being searched to find ways to reinvigorate the immune response. Combination strategies are aimed to eliminate the immunosuppressive microenvironment with the major goal of eliciting synergistic antitumor effects. Many clinical trials are evaluating the combined effect of anti-PD-1/PD-L1 therapy with other treatments, including chemotherapy, radiotherapy, other immunotherapeutic approaches or targeted therapies.7 One of the drugs that has been shown to modulate the TME, turning it into immunoresponsive is dasatinib, a multikinase inhibitor that targets SRC family kinases (SFKs) and BCR/ABL kinases and is approved for the treatment of chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL).8 In patients with leukemia, dasatinib has been shown to cause clonal expansion of CD8+ T lymphocytes and natural killer (NK) cells.9 10 Although dasatinib as a single agent showed modest clinical efficacy in patients with NSCLC in comparison with those undergoing chemotherapy, marked activity or stable disease was found in 11.7% patients, suggesting that there is a potential subpopulation of patients with high sensitivity to this drug.11 In preclinical murine models of melanoma, sarcoma, breast and colorectal cancer, dasatinib has been shown to cause antitumor effects by increasing the number of tumor-infiltrating CD8+ cells and decreasing that of Tregs.12 In addition, in DDR2-expressing syngeneic mouse models, dasatinib demonstrated synergistic effects with anti-PD-1 therapy.13 Nonetheless, the effect of dasatinib in NSCLC, in combination with immunotherapy, has not been explored.
Beyond the modulatory effect on the TME, it is well established that dasatinib also exerts remarkable direct effects on cancer cell growth in many solid tumors. In these malignancies, dasatinib inhibits the activity of various SFK members, such as SRC, YES, FYN and LYN, thereby inducing tumor cell cytotoxicity and apoptosis.8 We have previously demonstrated in NSCLC that dasatinib has a potent antiprimary tumor and antimetastatic activity in xenografts established with human cell lines and patient-derived xenografts (PDXs) that specifically express high levels of YES1.14 These data strongly support that YES1 could emerge as an appropriate biomarker of response to dasatinib in NSCLC.
We herein demonstrate that YES1 expression is found, among other SFK members, as the strongest predictor of poor prognosis in patients with NSCLC, and that high YES1 protein levels are associated with increased number of tumor-infiltrating Tregs. We also show in two different and clinically relevant NSCLC mouse models that the combination of dasatinib with anti-PD-1 has synergistic antitumor effects, leading to tumor regressions. Such effects are accompanied by a reduction in the number of Tregs within the tumors, which is likely due to an inhibition of Treg conversion and proliferation elicited by dasatinib, as we show here in vitro.
Materials and methods
Cohort of patients with NSCLC used for immunophenotyping and immunohistochemistry
Surgical samples from primary lung cancer were obtained from University Clinic of Navarra (CUN) (Pamplona, Spain). We included untreated patients diagnosed with NSCLC, with complete resection of the primary tumor after surgery. Tumors, classified according to the World Health Organization 2004 system, were stratified in agreement with the eighth TNM edition.15 The cohort included 116 patients diagnosed from 2000 to 2013. Reported Recommendations for Tumor Marker Prognostic Studies criteria were followed.16 Detailed clinical and pathological information of the cohort is summarized in online supplemental table 1.
Supplemental material
Multiplexed immunophenotyping and immunohistochemistry
Paraffin-embedded tissue microarrays (TMAs) containing three representative tissue cores per case were built and sectioned. For the multispectral immunophenotyping of human CD8, CD4, FOXP3 and DAPI (for nuclear staining) in the TMAs, we used a validated kit from Akoya Biosciences (Marlborough, Massachusetts, USA), according to the manufacturer’s recommendation. For multispectral immunophenotyping in mouse tumors, the murine-specific kit from Akoya (NEL840001KT) was used following the manufacturer’s instructions, with some modifications and additional markers. The kit includes the Alexa Fluor tyramides Opals 520, 570 and 690, as well as spectral DAPI. Opals 540 (FP1494001KT), 620 (FP1495001KT) and 650 (FP1496001KT) were not included in the kit and were purchased from Akoya. The following primary antibodies were used: anti-FOXP3, anti-CK, anti-CD31, anti-CD4, anti-CD8 and anti-F4/80. Antibodies and experimental details of human and mouse immunophenotyping can be found in online supplemental table 2. The sample scanning, spectral unmixing and quantification of signals were conducted with the Vectra Polaris Automated Quantitative Pathology Imaging System (Akoya), using the Phenochart and InForm V.2.4 softwares (Akoya). Data were given as number of cells with a specific immunophenotype/total number of cells.
For the detection of YES1 in the TMAs by immunohistochemistry, antigen retrieval was performed by heating the samples in citrate buffer (10 mM, pH 6), and the primary anti-YES1 antibody (Proteintech, 20 243–1-AP) was incubated at 1:100 dilution. For quantification, slides were scanned with the Aperio CS2 scanner (Leica, Barcelona, Spain) and images were visualized with the Aperio Image Scope V.12.1.05029. The H-score was then calculated following previously published protocols.17
Cohort of patients with NSCLC to study expression of SFK members: bioinformatic analysis
The description of the cohort and the bioinformatic analysis are described in online supplemental material and methods.
Supplemental material
Tumor microenviroment profiling in patients with NSCLC from TCGA using CIBERSORT
The details of the methodology used are described in online supplemental material and methods.
Cell lines
Previously characterized murine cell lines used for functional experiments were 393P18 19 and UNSCC680AJ (from now on referred to as UN680).20 393 P cells were a generous gift from Dr JM Kurie (The University of Texas MD Anderson Cancer Center, Houston, Texas, USA). cDNA from 393P, UN680, Lewis lung carcinoma (3LL)21; Lacun2 and Lacun322; and UNSCC679AJ (UN679 for simplification)20 was obtained in our Laboratory. cDNA from 334SQ, 389N1, 368T1, 389T2, 482N1, 482T1, 802T4, LKR10, LKR13, and LSZ223 was kindly donated by Dr Silvestre Vicent (CIMA). Cells were grown in RPMI-1640 (Gibco) or DMEM (Gibco) supplemented with 10% of HyClone Serum (Thermo Scientific), 1% penicillin–streptomycin (Lonza), at 37°C in 5% CO2 humidified atmosphere.
Cytotoxicity assay
The protocol of the assay is explained in online supplemental material and methods.
Real-time quantitative PCR (RT-qPCR) and western blotting
Primers and antibody specifications are shown in online supplemental tables 3 and 4, and the methodology for both techniques is described in online supplemental material and methods.
YES1 knockdown in the 393P cell line
The short hairpin RNA (shRNA) strategy, reagents and protocols are shown in online supplemental material and methods.
Mouse models
All animal experiments were approved by the Animal Committee of the University of Navarra. Murine adenocarcinoma 393P (4×106 cells) or squamous cell carcinoma UN680 (2×106 cells) cell lines were subcutaneously injected in one flank of 8-week-old female Sv/129 (Envigo) or A/JOlaHsd (Harlan-Winkelmann) mice, respectively. When tumor volume reached approximately 75 mm3, mice were randomized into four groups (eight per group) and treated with 30 mg/kg of dasatinib (Bristol-Myers Squibb) per day by oral gavage or vehicle (80 nM citric acid, pH 2.1; Sigma-Aldrich). The mouse-specific anti-PD-1 blocking antibody (RMP1-14, BioXCell) was administered the first day of dasatinib treatment (day 0) and at days 3 and 6 (100 µg per mouse, intraperitoneally). Tumor volume was measured with a digital caliper every 2 or 3 days and was calculated using the formula volume=(length×width2)/2.
A rechallenge experiment was carried out 2 months after tumor disappearance (seven mice from the combined group and five from the dasatinib group). Mice were subcutaneously injected with 393 P cells (2×106 cells) in the opposite flank, where the tumor was induced the first time. Six mice were injected for the first time as control group.
The effect of knocking down YES1 in tumor cells, in combination with anti-PD-1, was studied in an in vivo experiment with 393P-shRNA-transduced cells. We subcutaneously injected 4×106 cells in 8-week-old female Sv/129 mice (Envigo) (n=8 per group). Anti-PD-1 was administered at days 6, 9 and 12. Tumor volumes were measured as previously explained.
Depletion of CD8 (anti-mouse CD8α, clone 2.43, BioXcell), CD4 (anti-mouse CD4, clone GK1.5, BioXcell) or NK (anti-mouse NK1.1, clone PK136, BioXcell) cells was achieved by intraperitoneal injection of 100 µg of antibodies each time (n=4 injections).
The antitumor effect of dasatinib (30 mg/kg) was also tested in mice devoid of T cells. To this aim, 3×1 06 393 P cells were inoculated in the flank of 8-week-old female athymic nude mice (Harlan-Winkelmann, eight mice per group).
Flow cytometry analysis
Sv/129 mice were inoculated with 4×1 06 393 P cells. At day 4, daily treatment with dasatinib was initiated (30 mg/kg) and anti-PD-1 (100 µg per mouse) was injected at days 4, 7 and 10 from cell inoculation. On day 14, tumors and spleens were surgically excised, mechanically disaggregated and digested with collagenase (400 MandL units/mL, Roche) and DNase (10 mg/mL, Roche). Next, erythrocytes were lysed (lysis buffer: 155 mmol/L NH4Cl and 10 mmol/L KHCO3) and cell suspensions were preincubated with a monoclonal antibody targeting mouse CD16/CD32 (15 min RT, FcBlock, 2.4G2; BD Pharmingen). Extracellular staining was performed by incubating with fluorochrome-conjugated antibodies (15 min at 4°C) diluted in FACS buffer (0.1% azide, 1% bovine serum albumin in phosphate-buffered saline). All antibodies used are shown in online supplemental table 5. For intracellular staining, cell suspensions were fixed and permeabilized according to manufacturer’s instructions (eBioscience) and then labeled with anti-FOXP3 antibody (30 min at 4°C). Samples were acquired using a FACSCANTO II flow cytometer. Data were analyzed with FlowJo software. The gating strategy for the flow cytometry analysis is shown in online supplemental figure 4. Depletion of CD8+, CD4+ or NK1.1+ cells was validated in blood and spleen samples from mice using flow cytometry, as described previously. The effect of treatments on circulating immune cell populations (CD45+, CD8+, CD4+, CD25+ and FOXP3+ cells) was evaluated at day 16 in an independent in vivo experiment (eight mice per group) after administration of dasatinib (30 mg/kg), anti-PD-1 (100 µg per mouse) or the combination of both drugs.
Supplemental material
The effect of dasatinib on interferon gamma (IFN-γ)-induced PD-L1 expression in 393P and UN680 tumor cells was studied by flow cytometry. Briefly, 50 000 cells were seeded and treated with dasatinib for 72 hours and murine IFN-γ (500 U/mL, #315–05, Pepotrech) during the 24 hours prior to sample acquisition. Cell staining was performed with an anti-mouse PD-L1 antibody, at 1:500 dilution (MIH5, Thermo Fisher).
IFN-γ-based enzyme-linked immunospot (ELISpot)
The detailed method is described in online supplemental material and methods.
Treg isolation and proliferation assay
Treg cells were isolated from murine spleens of the previously characterized B6-Foxp3EGFP/B6.Cg-Foxp3tm2(EGFP)Tch/J (Foxp3GFP for short) reporter mice24 (Jackson Labs, stock #006772) using a FACSAria-Ilu cell sorter. After 3 hours of resting, 4×104 Treg cells were seeded in 96-well plates in the presence of plate-bound anti-CD3 (1 µg/mL, BD Pharmingen) and anti-CD28 antibodies (0.5 µg/mL, BD Pharmingen), IL-2 (100 U/mL) and dasatinib (2 and 10 nM). After 48 hours of incubation with dasatinib, Tregs were fixed, permeabilized and stained with anti-ki67 antibody (1:400, clone: 16A8, BioLegend). Dead cells were identified with Zombie NIR dye (1:2000, BioLegend) and were excluded from the analysis.
For the study of Treg cell signaling by western blotting, 7.5×104 Foxp3GFP+ cells were incubated with 10 nM of dasatinib for 45 min. Antibodies and their specifications of use are shown in online supplemental table 4. IL-10 levels were measured in cell supernatants using the mouse IL-10 ELISA Set, OptEIA (BD Biosciences).
CD4 purification and conversion into Treg cells
The effect of dasatinib on the conversion of CD4+ T lymphocytes into Treg cells was determined by flow cytometry and by western blotting. For this purpose, purified CD4+ cells were isolated from spleens of the Foxp3GFP reporter mice using a CD4+ T-cell isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. 6×104 (for flow cytometry) or 1×106 (for western blot) CD4+ T cells were seeded in the presence of anti-CD3 and anti-CD28 antibodies, IL-2 (as described previously), transforming growth factor beta (TGF-β) (5 ng/mL, Peprotech) and dasatinib (2, 10 and 20 nM). For flow cytometry analysis, T cells were stained with anti-CD4 (1:500, RMA4-5, BioLegend) and Zombie NIR dye. Antibodies used for western blotting are specified in online supplemental table 4.
Statistical analysis
Comparisons between two groups were performed using Student’s t-test when data followed a normal distribution or Mann-Whitney U test as non-parametric assay. Differences between more than two groups were analyzed with one-way or two-way analysis of variance followed by Bonferroni post hoc test. Statistical tests are specified in each figure. For Kaplan-Meier analysis using the publicly available datasets, the median cut-off value was used to stratify patients in high versus low levels. The log-rank test was used to calculate the statistical differences between Kaplan-Meier curves.
Data were analyzed with GraphPad Prism V.5 software (GraphPad). Statistical significance was defined as p<0.05 (*), p<0.01 (**) and p<0.001 (***).
Results
High YES1 expression is associated with infiltration of Tregs and predicts poor prognosis in patients with NSCLC
We first studied mRNA levels of the SFK members expressed in solid tumors (YES1, SRC, FYN and LYN) and compared levels between normal lung tissues and NSCLC specimens in the TCGA cohort. YES1 and SRC levels were significantly higher in NSCLC as compared with non-malignant tissues (figure 1A). By contrast, no differences were found for LYN, and lower expression of FYN was observed in tumors than in normal lung (figure 1A). We next assessed the prognostic significance of each SFK member individually or as an SFK signature using the Kaplan-Meier plotter, stratifying the patients by the median expression value. Survival analysis showed that high YES1 levels were significantly associated with shorter overall survival (OS) (HR 2, 95% CI 1.25 to 3.23; p=0.0034; figure 1B). High LYN levels were similarly associated with reduced OS, although with less prognostic value than that of YES1 (HR 1.62, 95% CI 1.02 to 2.58; p=0.039) (online supplemental figure 1A). SRC expression was not related to prognosis and low FYN levels predicted worse outcome (not shown). The signature including all these SFKs was also weakly but significantly associated with worse OS (HR 1.62, 95% CI 1.02 to 2.57; p=0.041) (figure 1C). Therefore, we conclude that YES1 is the strongest predictor of reduced OS in NSCLC, among all these SFKs.
Supplemental material

(A) Comparison of YES1, SRC, FYN and LYN mRNA expressions between non-malignant lung tissue and NSCLC (TCGA cohort). Mann-Whitney U test was used for the statistical analysis. (B,C) Kaplan-Meier survival curves showing that high YES1 and SFK levels (above the median) are associated with worse OS in NSCLC. The log-rank test was used for the statistical comparisons. (D,E) Quantification of FOXP3+CD4+ cells in NSCLC specimens from the CUN cohort. The percentage of Tregs (FOXP3+CD4+) in YES1-positive tumors in the upper quartile of the YES1 H-score is significantly higher than that found for the rest of the tumors (D) and in the lower quartile (E). (F,G) Quantification of FOXP3+CD4+ cells in patients with LUAD from CUN. The percentage of Treg cells in the YES1 upper quartile was compared with the rest of the quartiles (F) or the lower quartile (G). (H) Representative images of YES1 IHC and multiplex immunofluorescence for CD8+, CD4+ and FOXP3+CD4+ cells in patients with NSCLC. (I) Relative abundance of Treg cells in patients with LUAD from the TCGA database analyzed with CIBERSORT tool. Scale bar: 50 µm. (D–G,I) Data are expressed as median, and statistical comparisons were performed using the Mann-Whitney U test. *P<0.05, **P<0.01. CUN, University Clinic of Navarra; IHC, immunohistochemistry; LUAD, lung adenocarcinoma; ns, not significant; NSCLC, non-small cell lung cancer; OS, overall survival; SFK, SRC family kinase; SRC, Treg, regulatory T cell.
Based on these results, we hypothesized that high YES1 levels could be associated with a more immunosuppressive TME, which could explain the worse outcome of patients with high levels of YES1. To address this, we used a series of 116 patients from CUN and quantified YES1 protein expression together with the immune markers CD4, CD8 and FOXP3. YES1 was evaluated by immunohistochemistry after calculation of the H-score, whereas the rest of the markers were analyzed by quantitative multiplex immunophenotyping using the Vectra Polaris System. The YES1 H-score was stratified by quartiles to classify patients into high (upper quartile) versus low (75% of patients below the upper quartile) expression. Interestingly, those patients with high YES1 levels showed also significantly higher number of tumor-infiltrating Tregs (FOXP3+/CD4+ cells) (p=0.041, figure 1D), suggesting that tumors with YES1 expression tend to be more immunosuppressive. When comparing the upper quartile versus the lowest quartile (25% of patients with the lowest YES1 expression), the differences in the number of Tregs were even more pronounced (p=0.009, figure 1E). Moreover, we found that this association occurred in patients with lung adenocarcinoma (LUAD) but not in those with lung squamous cell carcinoma (LUSC) (figure 1F,G and online supplemental figure 1B,C). On the contrary, no relationship between expression of YES1 and CD4+/FOXP3− or CD8+ cells was found (online supplemental figure 1D,E). Representative images of tumors with high versus low YES1 levels and high versus low number of CD4+/FOXP3+ cells are shown in figure 1H.
In order to validate these results, we performed an in silico analysis using data from TCGA and CIBERSORT,25 a computational tool that infers the presence of specific immune cell populations in a given patient based on gene expression tumor profiles associated with a specific immune cell type. This analysis showed that patients with LUAD with high YES1 expression tended to have a higher number of Treg cells, although this difference did not reach statistical significance (p=0.055, figure 1I). There was no association between YES1 expression and the number of Treg cells in LUSC (online supplemental figure 1F).
SKF expression in murine NSCLC cancer cell lines and in vitro effect of dasatinib
Then we sought to select immunocompetent NSCLC mouse models that were appropriate to investigate the immunomodulatory effect of dasatinib in vivo, with or without combination with anti-PD-1 therapy. Recent studies have shown the direct antiproliferative effect of this SFK inhibitor on tumor cells, as well as its role in altering the immunosuppressive TME.12 In addition, we have previously shown that YES1 is an accurate predictor of response to dasatinib in NSCLC.14
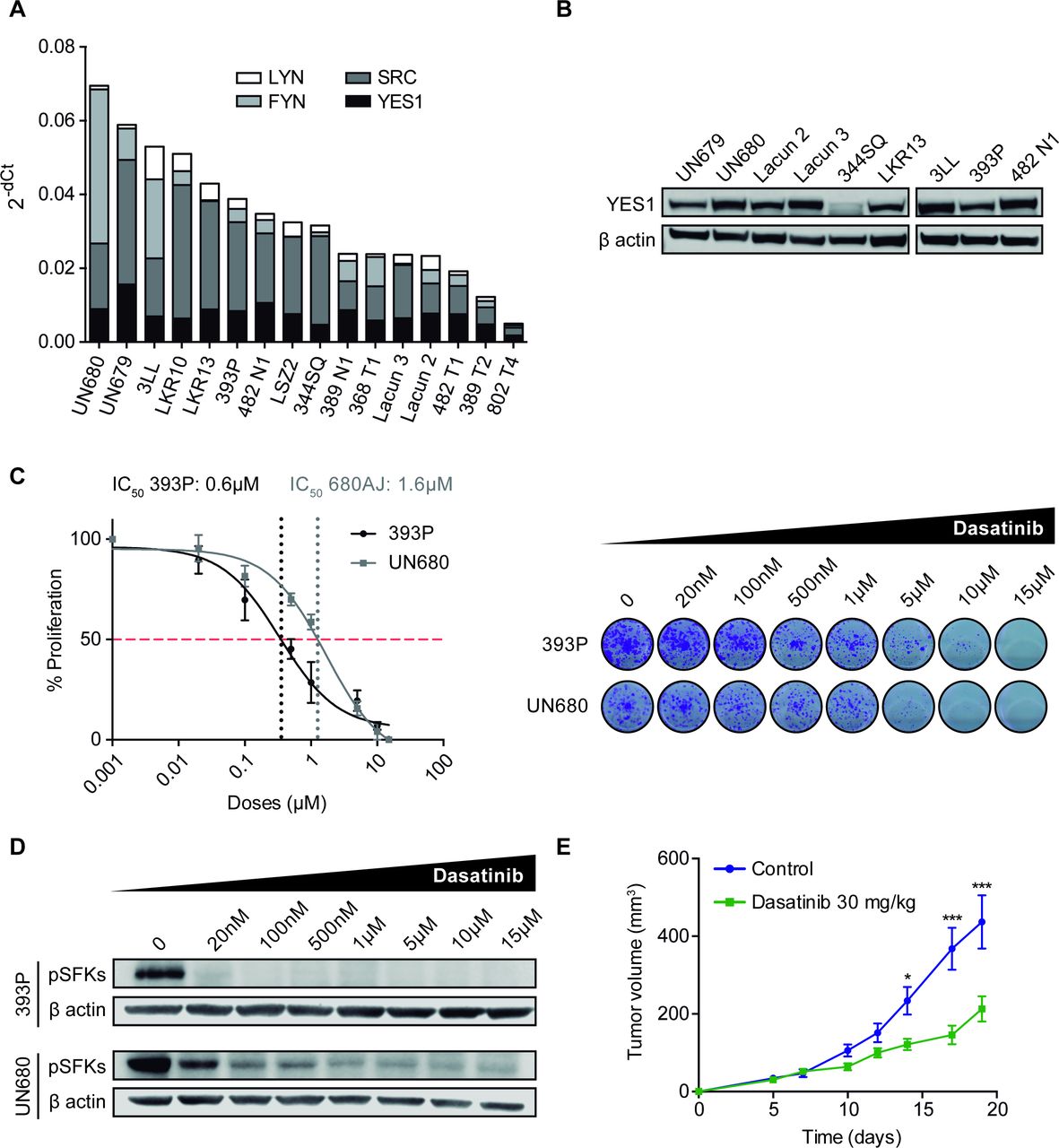
We screened a panel of murine NSCLC cell lines (n=16), including adenocarcinoma and squamous cell carcinoma histologies and quantified mRNA levels of the SFK family members. RT-qPCR revealed that most cell lines showed expression of YES1, SCR, FYN and LYN, although with different mRNA levels (figure 2A). We also confirmed by western blotting the protein expression of YES1 in some of the cell lines (n=9) (figure 2B) and selected 393P and UN680 cells for further experiments, due to high YES1 levels and their response to anti-PD-1 therapy in vivo, according to our previous results.19 20
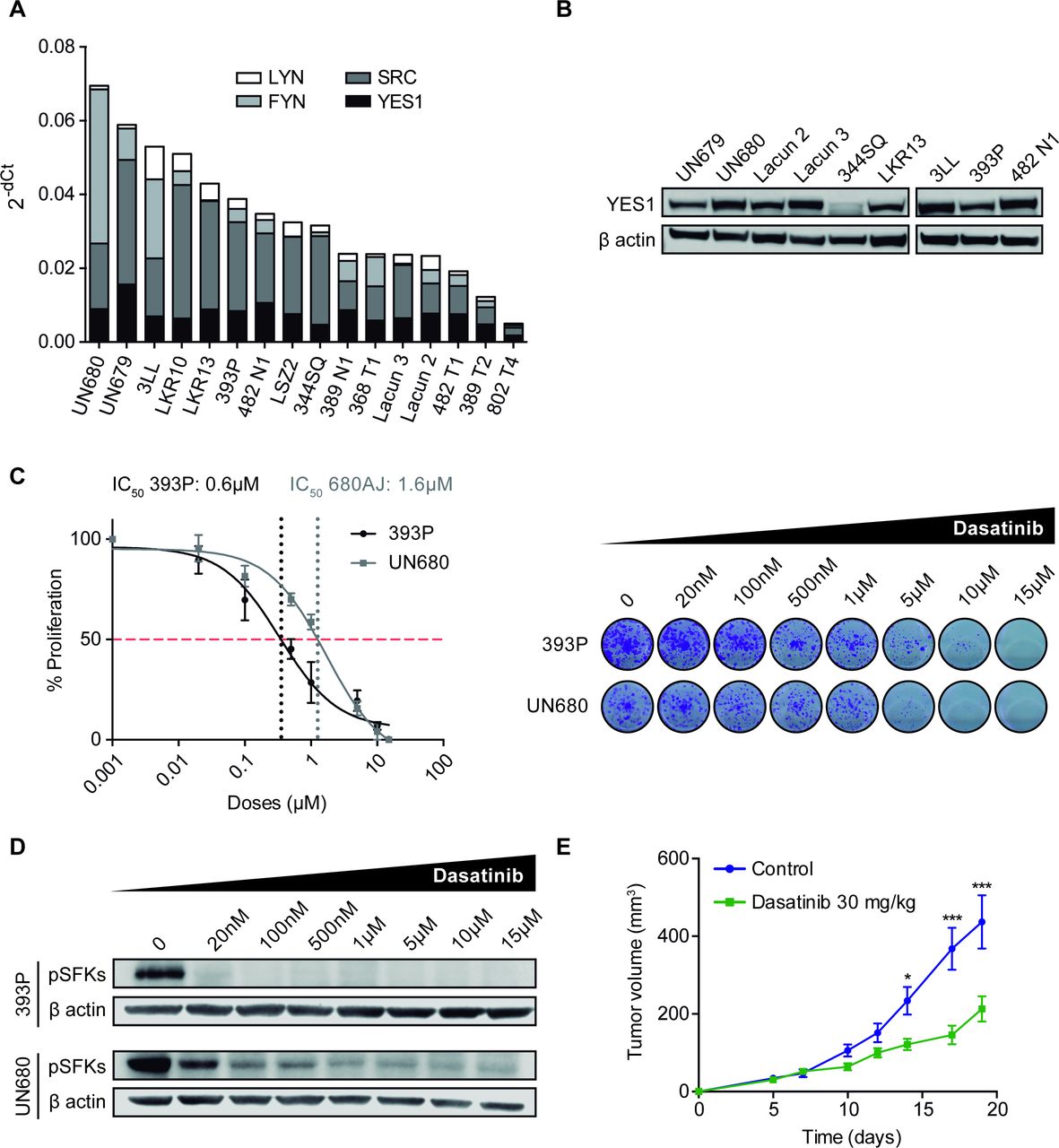
(A) mRNA expression of YES1, SRC, FYN and LYN analyzed by RT-qPCR in a panel of 16 murine NSCLC cell lines. (B) Western blot analysis of YES1 expression in nine murine cell lines. (C) Effect of dasatinib on 393P and UN680 cell proliferation in vitro. (D) Western blot analysis showing pSFK protein inhibition by dasatinib at 10 hours post-treatment in vitro. (E) Subcutaneous tumor growth of 393 P cells injected in athymic nude mice treated with dasatinib (30 mg/kg) or vehicle. A two-way analysis of variance followed by a post hoc Bonferroni test was used. *P<0.05, ***P<0.001. NSCLC, non-small cell lung cancer; pSFK, phospho-SRC family kinase; RT-qPCR, real-time quantitative PCR; SFK, SRC family kinase.
The direct effect of dasatinib on proliferation was then evaluated in vitro for 393P and UN680 cells. Both cell lines responded to dasatinib in a dose-dependent manner (figure 2C) with an IC50 of <2 µM, similar to what has been described for SFK-expressing human cancer cells.14 On-target specificity of dasatinib treatment was verified by western blot analysis of SFK phosphorylation. As shown in figure 2D, a decrease in phospho(p)-SFKs was observed on dasatinib administration (20 nM–15µM). Of note, there is currently no antibody to detect specifically the phosphorylated form of YES1. As dasatinib may also affect the lymphocyte-specific protein tyrosine kinase (LCK) of T cells,26 which can partially mediate its antitumor effects, we performed an in vivo experiment where 393 P cells were injected into athymic nude mice (lacking T cells) that were treated with 30 mg/kg dasatinib. Results showed that dasatinib exerted a significant therapeutic effect, with a 51% reduction in tumor volume compared with controls (figure 2E and online supplemental figure 1G).
Dasatinib synergizes with anti-PD-1 immunotherapy in NSCLC causing tumor regression
The in vivo effect of dasatinib alone or in combination with anti-PD-1 was then assessed in the two immunocompetent NSCLC models. In the 393P subcutaneous adenocarcinoma model, treatment with dasatinib resulted in a highly significant reduction in tumor volume (~70% at day 35 postcell injection with respect to controls), whereas anti-PD-1 showed a modest decrease (~24%) (figure 3A). The combination of dasatinib with anti-PD-1 caused a dramatic reduction of tumor growth (p<0.001), with seven out of eight complete tumor rejections (figure 3A,B). OS of mice was significantly prolonged in dasatinib-administered mice, but was increased much further for the combination group, with seven animals (87.5%) alive at the end of the study (100 days after cell injection) (figure 3C).

(A) Subcutaneous tumor volume in the 393P model (Sv/129 mice) treated with dasatinib (30 mg/kg/day), anti-PD-1 (100 µg, days 7, 10 and 13), or both (n=8). Statistical differences are indicated for the comparisons between double-treated and control groups. (B) Waterfall plot showing the individual tumor volume changes between days 7 and 35 in the 393P model. (C) Survival curves of the in vivo experiment shown in (A). Log-rank test was used for statistical analysis. A final volume of 1000 mm3 was used as endpoint criteria. (D) Rechallenge experiment using 393 P cells injected in tumor-rejected mice, 60 days after tumor disappearance. Naïve mice were used as control group (n=6). (E) Analysis of 393P-specific IFN-ɣ secretion by splenocytes isolated from the mice of the rechallenge experiment. IFN-ɣ levels were measured using ELIspot technique. Student’s t-test was used for comparison. (F) Tumor growth of UN680 cells implanted in A/JOlaHsd mice and treated with dasatinib (30 mg/kg, daily), anti-PD-1 (100 µg, days 5, 8 and 11), or both (n=8). (G) Waterfall plot of the volume changes in UN680 tumors between days 5 and 16. (A,F) Data are expressed as mean±SEM and were analyzed with a two-way analysis of variance followed by a post hoc Bonferroni test. Statistical differences show the comparison between controls versus the double-treatment group. *P<0.05, **P<0.01, ***P<0.001. ELIspot, enzyme-linked immunospot; IFN-γ, interferon gamma; PD-1, programmed cell death 1.
A rechallenge experiment was performed 60 days after tumor rejections to test immunological memory. All mice coming from anti-PD-1 (n=1), dasatinib (n=4) and dasatinib+anti-PD-1 (n=7) groups were refractory to the development of new tumors (figure 3D, anti-PD-1 not shown). Then, an ELIspot assay was carried out by culturing splenocytes from these mice with irradiated 393 P cells. As shown in figure 3E, IFN-γ secretion by T cells was significantly higher (p<0.05) in the dual-treatment group compared with anti-PD-1 or dasatinib alone, suggesting a more effective long-lasting memory response when combining dasatinib and anti-PD-1. Tumor growth for each one of the animals in shown in online supplemental figure 1H.
Assessment of pSFK levels and some of the downstream molecules related to dasatinib activity was carried out by western blot analysis in tumors from the different experimental groups in an independent short-term treatment in vivo experiment (see further). As shown in online supplemental figure 2A,B, levels of pSFK were strongly reduced in the combination group, with a more modest decrease in the single-treatment groups. No changes were observed for the other phosphoproteins tested: pAKT, pSTAT3 and pERK1/2.
Supplemental material
We also studied the effect of this drug combination in an alternative NSCLC model: a squamous cell carcinoma syngeneic mouse model developed by subcutaneous injection of UN680 cells, previously developed and characterized in our laboratory.20 Although the combinatory effect was less pronounced in this model, results were similar to the 393P model, with therapeutic improvement for the combination between dasatinib and anti-PD-1 (figure 3F,G and online supplemental figure 1I). In this case, one tumor regression was achieved in the experiment, corresponding to the combination group (figure 3G).
No weight loss or external signs of toxicity, such as lordosis or distress, were observed in the single-treated or combined-treated groups of animals.
To ascertain to what extent expression of YES1 in cancer cells was mediating the therapeutic effect, we silenced YES1 in 393 P cells using shRNA and performed an in vivo experiment with or without combination with anti-PD-1 (online supplemental figure 2C,D). As expected, YES1 knockdown improved anti-PD-1 efficacy, obtaining a 52% reduction in the tumor volume for the shYES1+ anti-PD-1 group compared with the untreated controls (sh-scramble) and 41% with respect to the sh-scramble +anti-PD-1 treated group (online supplemental figure 2C,D).
Combination of dasatinib and anti-PD-1 reduces Treg cells in the TME and blood
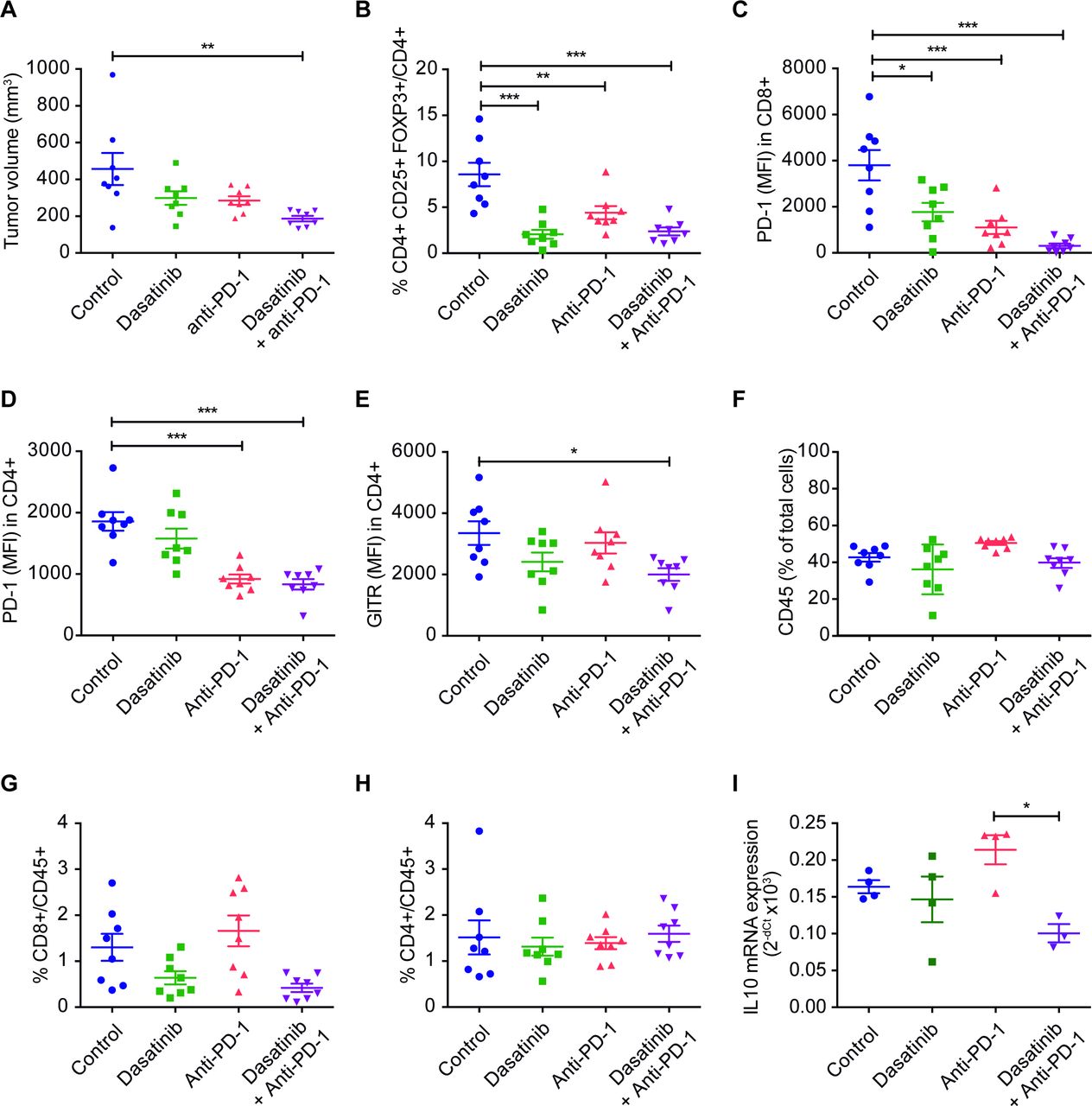
Changes in the tumor immune infiltrate for the different experimental groups were assessed by flow cytometry in the 393P model in an additional (short-term treatment) in vivo experiment, where tumor samples were obtained at day 14, time at which tumor volume curves from the different groups begin to separate (figure 4A and online supplemental figure 3A). Dasatinib and/or combination treatments led to a very significant decrease in the number of Tregs (CD4+CD25+FOXP3+) and levels of PD-1 in CD8+ and in CD4+ cells (the latter ones likely reflecting exhausted T lymphocytes) (figure 4B–D). We also observed significantly lower levels of GITR (another marker of Tregs) in CD4+ cells, but only in those mice treated with dual blockade (figure 4E). None of the treatments alone or in combination modified the frequency of CD45+, CD4+, CD8+, NK cells or macrophages (figure 4F–H and online supplemental figure 3B–I). The gating strategy followed for the analysis of lymphocyte subpopulations can be found in online supplemental figure 4. Levels of the Treg-associated cytokine IL-10 were also significantly reduced in tumors from animals administered with the drug combination (figure 4I). In addition, we studied whether dasatinib modified PD-L1 levels in either 393P or UN680 cells in vitro, but no changes were found with respect to controls (online supplemental figure 3J,K).
Supplemental material

(A) Tumor volumes of 393 P cells, at the moment of conducting the flow cytometry assay (day 14). Mice were treated with dasatinib (30 mg/kg/day), anti-PD-1 (100 µg, days 4, 7 and 10), or both (n=8). (B–H) Percentage of cells or MFI of tumor immune populations: tumor-infiltrating Treg (FOXP3+CD25+CD4+) (B), PD-1 in CD8+ (C), PD-1 in CD4+ (D), GITR in CD4+ (E), CD45+ (F), CD8+/CD45+ (G) and CD4+/CD45+ (H) cells. (I) mRNA expression of IL-10 in 393P tumors. Data are represented as mean±SEM. Comparisons were analysed using a one-way analysis of variance followed by a posthoc Bonferroni test. *P<0.05, **P<0.01, ***P<0.001. IL, interleukin; MFI, median fluorescence intensity; PD-1, programmed cell death 1; Treg, regulatory T cell.
In order to further investigate changes in the tumor immunolandscape due to the different treatments, we performed in situ multiplex immunofluorescence quantification of CD4, CD8, F4/80, CD31 and FOXP3 infiltrating cells in 393P tumors. Interestingly, consistent with the flow cytometry results, the combined treatment dasatinib+anti-PD-1 led to a significant decrease in the number of Treg cells (figure 5A). Representative images of FOXP3+/CD4+ cells are shown in figure 5B and a representative picture of a wide-field tumor where all seven markers are merged, in figure 5C. A lower number of CD31+ cells was also detected in animals administered with anti-PD-1 and the combination treatment, suggesting an impaired angiogenesis in these groups. No changes with respect to controls were observed for the other cell populations (online supplemental figure 5A–E).
Supplemental material

(A) Quantification of cells coexpressing FOXP3 and CD4 (Treg cells) in 393P tumor microenvironment (n=8) analyzed by mIF. (B) Representative mIF images showing Treg cells in each group. (C) Example of an mIF landscape image showing CK+, CD8+, CD4+, F4/80+, CD31+ and FOXP3+ cells in one of these tumors. (D) 393P tumor volumes at the time (day 16) of the characterization of circulating immune cells by flow cytometry. (E–G) Percentage of Tregs (E), CD8+ (F) and CD4+ (G) cells in blood. Differences were analyzed with one-way analysis of variance followed by a post hoc Bonferroni test. *P<0.05, **P<0.01, ***P<0.001. mIF, multiplex immunofluorescence; PD-1, programmed cell death 1; Treg, regulatory T cell.
We next evaluated whether dasatinib alone or in combination with anti-PD-1 would not only decrease the infiltration of Tregs in the TME but also deplete circulating Tregs. For this purpose, we quantified CD4+, CD8+ and CD4+/FOXP3+/CD25+ cell populations in blood, in an independent in vivo experiment, at day 16 (figure 5D–G and online supplemental figure 5F). Interestingly, Treg cells were prominently reduced in the dasatinib and the combination groups (figure 5E). No changes were found in the number of Tregs in anti-PD-1 treated mice, which shows that the depletion of these cells is mainly mediated, at least in blood, by dasatinib monotherapy. Levels of CD4 and CD8 T cells remained unchanged with any of the treatments (figure 5F,G).
CD4+ and CD8+ T-cell depletions diminish the effect of dasatinib and anti-PD-1 on tumor growth
To assess which immune populations were involved in the response to dasatinib and anti-PD-1 in vivo, we selectively depleted CD4, CD8 or NK lymphocytes in mice bearing 393P tumors (figure 6A,B). T cells and NK cells were depleted 24 hours prior to the treatment initiation. Levels of cell depletion were tested in blood and spleen by flow cytometry at the end of the experiment. An example of the drop in CD8+ population in treated animals is shown in online supplemental figure 6A. All groups responded to the combination treatment compared with controls until day 12. This delay in tumor growth could be associated with the direct effect of dasatinib on tumor cell proliferation. However, from that time point on, dasatinib and anti-PD-1 effectiveness was significantly abrogated in CD4-depleted and CD8-depleted mice, demonstrating that these subpopulations of cells are required for the antitumor activity of the combined therapy (figure 6A,B).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Schematic representation of the experimental design followed for the depletion of CD8, CD4 and NK1.1 cell populations and the treatment with dasatinib+anti-PD-1 in vivo. (B) Tumor growth of 393P-inoculated cells in the presence of depleting antibodies and treatment with dasatinib+anti-PD-1 (n=6). (C) Treg cell proliferation 4 days after plating in the presence of IL-2, anti(α)CD3, αCD28 and dasatinib (2 and 10 nM) measured by flow cytometry. (D) IL-10 levels measured in Treg cells culture medium after 48 hours of exposure to dasatinib. (E) Protein expression of pLCK, LCK, YES1, pSTAT3, STAT3, pSTAT5 and STAT5 in Treg cells treated with dasatinib (10 nM) for 45 min. (F,G) In vitro experiment of TGF-β-dependent CD4+ T cell conversion into Tregs, in the presence of IL-2, αCD3, αCD28 and dasatinib (2, 10 and 20 nM), analyzed by flow cytometry. (H) Western blotting of pSTAT5, STAT5, pSMAD3 and SMAD3 in CD4+ T cells converted into Tregs after treatment with dasatinib (10 nM). Differences between groups were evaluated with a two-way (B) or a one-way analysis of variance test (C,D,F) followed by a post hoc Bonferroni test. **P<0.01, ***P<0.001, ****P<0.0001. IL, interleukin; LCK, lymphocyte-specific protein tyrosine kinase; PD-1, programmed cell death 1; pLCK, phospholymphocyte-specific protein tyrosine kinase; TGF-β, transforming growth factor beta; Treg, regulatory T cell.
Dasatinib impairs proliferation and inhibits LCK phosphorylation in Treg cells
We investigated whether dasatinib could exert an antiproliferative effect on Treg cells, as seen in tumor cells. Dasatinib effectively diminished Treg proliferation at very low doses (2 and 10 nM) (figure 6C). This decrease was accompanied by a very significant reduction in IL-10 levels released by these cells (figure 6D). We next assessed the molecular mechanism behind the effect of dasatinib on Treg cell proliferation. To this aim, we studied activation of the LCK, a direct target of dasatinib, whose phosphorylation at Y394 is necessary for TCR signaling initiation.27 A strong inhibition of pLCK levels was found in the Treg cells treated with dasatinib (10 nM) compared with untreated cells (figure 6E). YES1 protein was also detected in Tregs, although levels were lower than those of LCK. Direct targeting on YES1 phosphorylation by dasatinib in Tregs could not be studied due to lack of specific anti-phosphoYES1 antibodies. Under these conditions, dasatinib did not alter pSTAT3 nor pSTAT5 levels in Treg cells (figure 6E and online supplemental figure 6C).
Dasatinib blocks CD4 T-cell conversion into Treg cells in vitro
We next evaluated whether the decrease in the percentage of Tregs observed in 393P tumors could be due not only to an inhibition of Treg cell proliferation but also to a blockade of the conversion of effector CD4+ T cells into Tregs. For this purpose, CD4+ CD25- cells were cultured with TGF-β to induce their differentiation into Treg cells (CD4+CD25+FOXP3+), according to standard protocols,28 in the presence of dasatinib. Remarkably, CD4+CD25- cell conversion was strongly reduced by dasatinib in a dose-dependent manner (figure 6F,G), suggesting that the drop observed in the number of tumor-infiltrating Tregs in the dasatinib+anti-PD-1 treatment is mediated by an impairment of CD4 T-lymphocyte conversion into Tregs and Treg cell proliferation. Levels of pSMAD3/total SMAD3 were subsequently evaluated, showing that dasatinib (10 and 20 nM) decreased the amount of both the phosphorylated and the total protein (figure 6H and online supplemental figure 6C–F). Moreover, dasatinib totally impeded the activation of STAT5 (figure 6H). Based on these results, we conclude that dasatinib inhibits TGF-β-dependent CD4 T-cell conversion by diminishing SMAD3- and STAT5-mediated signaling.
Legends of the supplemental figures can be found in online supplemental file 3.
Supplemental material
Discussion
Research aiming to overcome primary and acquired resistance against anti-PD-1 therapy in patients with NSCLC and with other malignancies is currently a priority. In fact, more than 1000 clinical trials are assessing combinations of immunotherapy (mainly anti-PD-L1/PD-1 or CTLA-4 antibodies) with other therapeutic strategies. Some approaches are using immunomodulatory drugs to unlock and reactivate the immune system.1 2 However, given the large amount and variety of such proposed drugs and strategies, the use of appropriate syngeneic animal models where these combinations can be easily tested offers a great tool that may guide clinical trials. Here, we report the successful combination between dasatinib (a drug approved by the FDA for leukemia) and anti-PD-1 for the treatment of NSCLC in two clinically relevant mouse models. This combination reduces the number of tumor-infiltrating Tregs, causing tumor regressions and inducing immunological memory, with long-lasting response in these animals.
An increasing number of studies is showing that the TME immunolandscape is determined by specific genetic alterations and expression of certain genes in cancer cells. For example, the coexistence of KRAS and STK11 (LKB1) mutations, which is estimated in 8%–30% of patients with NSCLC, is associated with an immunosuppressive TME characterized by high infiltration of tumor-associated neutrophils that secrete IL-6 and CXCL-10.29 30 On the contrary, concurrent KRAS-TP53 mutations are associated with infiltration of effector CD8+ T lymphocytes and responsiveness to anti-PD-1 therapy.31 Tumor cell-intrinsic activation of β-catenin signaling has been linked to a non-T-inflamed TME and resistance to anti-PD-1 therapy.32 Similarly, gain in Myc function inhibits CD4+ T-cell activation and infiltration.33 34 Here we show that expression of the SFK member YES1 in ADC is significantly associated with an increase in the number of Tregs in patients with NSCLC. This suggests that YES1 induces an immunosuppressive TME that could hinder the effect of anti-PD-1 therapy. Among the SFK members commonly expressed in NSCLC, high levels of both YES1 and LYN predicted reduced OS in patients with NSCLC, with YES1 being clearly the most potent predictor of poor prognosis. This is in agreement with a previous report from our group showing that YES1 overexpression and gene amplification were associated with a reduction in both relapse-free survival and OS.14 In our lung cancer models, we have shown, using shRNA strategies, that abrogation of YES1 in tumor cells is responsible for ~50% of tumor reduction when combined with anti-PD-1.
Among the drugs that inhibit the SFKs, dasatinib has been tested in clinical trials for NSCLC11 and other solid tumors. This multityrosine kinase inhibitor is currently approved for the treatment of CML and ALL, while its effectiveness alone or in combination with other drugs in non-hematological malignancies is still under investigation. A fundamental issue in NSCLC is to accurately define biomarkers of drug response, since clinical benefit has been found to be restricted to a small percentage of patients.5 Huang et al35 identified a six-gene signature that predicted sensitivity to dasatinib in lung cancer cell lines, although such putative biomarkers have not been assessed in patients yet. Dasatinib causes cell growth inhibition and apoptosis in NSCLC cells with high expression of YES1, whereas in low-expressing or negative cells, the effect is much less pronounced.14 Moreover, a potent antitumor effect has been demonstrated in tumor growth and metastasis of cell line-derived and PDXs that express high YES1 levels.14 Therefore, YES1 is postulated as a biomarker of response to dasatinib in NSCLC. In addition, based on results presented here, effectiveness of dasatinib could be monitored in blood as a reduction in the number of Treg cells.
Dasatinib exerts direct effects on both cancer cells and cells of the TME, as results presented here and reports in immunocompetent models of melanoma, sarcoma, breast and colorectal cancer have shown.12 Mechanistically, the antitumor activity observed in these models was related to an increase in the number of infiltrating CD8+ cells and reduction of Tregs.12 Tu et al13 have recently reported that combination between dasatinib and anti-PD-1 results in a synergistic antitumor growth in DDR2-expressing murine colon cancer and sarcoma models. In agreement with these results, our study, using dasatinib alone or in combination with anti-PD-1, has demonstrated a very significant drop in the number of Tregs and exhausted CD8+ and CD4+ T lymphocytes, especially for the drug combination. This is also concomitantly accompanied by a decrease in intratumoral levels of IL-10, a cytokine released by Tregs with tolerogenic and anti-inflammatory properties that dampens immune responses.36 IL-10 signaling is required for the immunosuppressive role of Tregs, altering the efficient activation of T cells.36 37 High levels of this cytokine in tumors and serum from patients with NSCLC have been found to be related with worse outcome.38 39
Depletion experiments revealed that both CD8+ and CD4+ T lymphocytes, but not NK cells, are required for the effective response to the combined therapy. Nonetheless, recovery of tumor growth after CD8+/CD4+ depletion was not fully achieved, which argues in favor of a direct effect of dasatinib on tumor cells, in addition to the effect on the T lymphocyte population.
Complex mechanisms may account for the intratumoral accumulation of CD4+/FOXP3+/CD25+ Tregs, including recruitment, expansion and differentiation from conventional effector CD4+ T lymphocytes.37 In our study, we hypothesized that dasatinib could alter proliferation and/or prevent the conversion from CD4+ cells into Tregs. We have proven the novel effect of dasatinib on alteration of Treg cell proliferation and conversion from CD4+ cells: a dose of as low as 10 nM dasatinib was able to reduce Treg proliferation by 96.3% and conversion from CD4 T cells by 76.7%. Effects were associated with reduction in levels of active LCK, STAT5 and SMAD3. Interestingly, Dyck et al40 have shown that anti-PD-1 inhibits Tregs conversion to unleash intratumoral effector T cells. Taken together, these data suggest that the main mechanism behind the effectiveness of combining dasatinib and anti-PD-1 therapies may rely on a full blockade of the Treg proliferation/conversion by both dasatinib and anti-PD-1, thus taking the brakes off the tumor specific cytotoxic effect of T lymphocytes.
Although the use of dasatinib in NSCLC alone or in combination with chemotherapy or EGFR inhibitors has been somewhat disappointing so far, our results provide the preclinical rationale for the combined use of dasatinib and PD-1/PD-L1 blockade to improve outcomes of patients with NSCLC. Of note, the clinical trial FRACTION (NCT02750514) is exploring combination between dasatinib and ICI in solid tumors.
Conclusions
Protein expression of YES1 is associated with poor prognosis and increased numbers of Tregs in patients with NSCLC. Dasatinib synergizes with anti-PD-1 to impair tumor growth in NSCLC experimental models and reduces the number of Tregs. CD8+ and CD4+ lymphocytes are necessary for the efficacy of the combination. The relevance of these results is that we provide a rationale for the combined use of dasatinib and anti-PD-1 to improve outcomes of patients with NSCLC.
Acknowledgments
We are grateful to Javier Garcia (University of Navarra) for technical help.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @FranExpositoR, @jose_lasarte
Contributors ER, IG, JA, LM, AC, JGP, JL, TL and RP participated in the design of the experiments and in the discussion of the results; ER, IG, YS, DA and SO participated in the flow cytometry experiments; MR, MV and CEDA worked on the multiplex analysis; ER, TL and JL were involved in experiments related to regulatory T cells; ER, IG, DS, AR, CS, and CB participated in the animal work; FE performed the bioinformatic analysis; ER and AC wrote the manuscript.
Funding This work has been funded by the Foundation for Applied Medical Research, ISCIII-Fondo de Investigación Sanitaria-Fondo Europeo de Desarrollo Regional 'Una manera de hacer Europa' (PI19/00230 to AC, PI19/00098 to LMM, PI17/00411 to RP and DA, PI19/00560 to JMGP; CIBERONC CB16/12/00443 to LMM and CB16/12/00390 to JMGP), AECC and Ramón Areces Foundations (both to LMM), 'Instituto de Investigación Sanitaria del Principado de Asturias to JMGP and BMS (to JA and LMM). Fellowship support: ER and YS, 'FPU, Spanish Ministry of Education'; IG, Gobierno Vasco; DS, 'Juan de la Cierva-Incorporacion, Spanish Ministry of Science and Innovation'; FE, 'Asociación de Amigos de la Universidad de Navarra', in association with 'La Caixa' Banking Foundation. We thank Dr JM Kurie (The University of Texas MD Anderson Cancer Center, Houston, Texas, USA) for the murine cell lines.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The ethical committee of the University of Navarra approved the study and signed informed consent was obtained from each patient. Experiments with animals were performed according the protocol approved by the ethical committee of the University of Navarra. The study was conducted according to the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.